A Chronicle of Survival: A Case Study of Chediak Higashi Syndrome with Good Outcome in Pakistan
Abstract
Chediak higashi syndrome is a unique hereditary disorder characterized by
oculocutaneous albinism, mild coagulation defects, progressive neurological
deficit, and an immense risk of developing hemophagocytic lymphohistiocytosis (hlh).
On that account, we report a case of a three-year-old child with chediak
higashi syndome in rawalpindi, pakistan. The diagnosis was established based on
the congregation of classical symptoms: ocular and cutaneous hypopigmentation,
repetitive infections, bleeding diathesis, and the pathognomic presence of
giant granules in white blood cells.
Though the prognosis of this disease is poor in the accelerated phase
with a high mortality rate, our patient upon timely diagnosis and hematopoietic
stem cell transplantation is living a healthy life now at the age of four
years
Keywords: chediak higashi
syndrome; hypopigmentation; bleeding diathesis; hemophagocytic
Introduction
Chediak higashi syndrome is a rare autosomal recessive
disorder with fewer than 500 cases reported throughout the world over the last
two decades1. In our literature
review, only 15 cases have been reported in pakistan to date among whom 7 died
during their first hospital admission. It is caused by a defect in the
lysosomal trafficking regulator gene lyst/chs1, leading to abnormal protein and
vesicle formation in all granule-containing cells of the body2. The syndrome is characterized by a triad of
oculocutaneous albinism, increased susceptibility to infections, and
coagulation defects3. A vast majority
of patients with chediak higashi syndrome develop an accelerated phase, named
hemophagocytic lymphohistiocytosis manifesting in the form of high-grade fever,
hepatosplenomegaly, pancytopenia and lymphohistiocytic infiltration of spleen,
liver and lymph nodes4. The presence
of giant cytoplasmic granules in white blood cells along with regularly
distributed clumped melanin along the entire hair shaft on hair microscopy
strongly proposes chediak higashi syndrome. Detection of a pathogenic variant
in the lyst/chs1 gene acts as a confirmatory marker for chediak higashi
syndrome5. Allogeneic hematopoietic
stem cell transplantation (hsct) is the treatment of choice. Based upon the
scarcity of this disease, the discrete clinical and hematological picture, and
a meager survival rate, we report a case of chediak higashi syndrome that was
treated successfully via hematopoietic stem cell transplantation.
Case presentation
The
patient was a 3-year-old female who presented with abdominal pain and
distention for 2 months along with 8-month history of recurring fever,
occasional epistaxis, and progressive pallor.
There
was a prior history of recurrent respiratory infections 5 times since birth
settled for the time being with antibiotics. She is the second child from a
consanguineous marriage with no family history of the disease.
Upon
examination, the patient was febrile, had golden grey hair and hypopigmented
skin (table 1). The patient had a
distended abdomen with moderate hepatosplenomegaly (figure 1).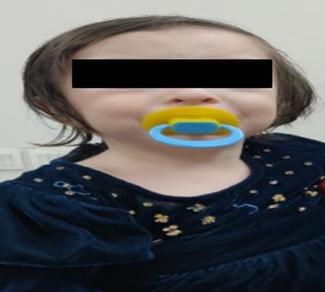
Figure 1: photograph of our patient showing golden grey hair
and hypopigmented skin.
Table 1:
laboratory investgations of our patient
|
Parameters |
Results |
Reference
values |
|
Haemoglobin |
7.9g/dl |
4-11g/dl |
|
Platelets |
20 × 109/l |
150-400 × 109/l |
|
Esr |
19mm/hr |
<15mm/hr |
|
Serum triglycerides |
3.33mmol/l |
1.7mmol/l |
|
Serum fibrinogen |
80mg/dl |
200-400mg/dl |
|
Serum ferritin |
579ng/ml |
7-140ng/ml |
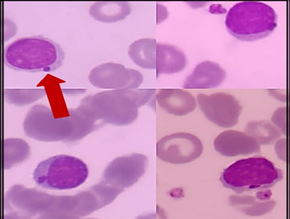
 (2a) (2b) (2c)
(2a) (2b) (2c)Figure 2: the giant azurophilic granules in white blood cells (red arrows)
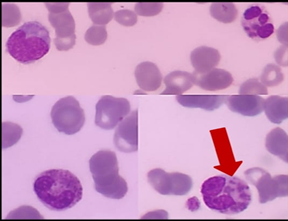
Bone marrow examination showed hypercellular fragments and giant cytoplasmic granules in all myeloid lineage. (figure 3).
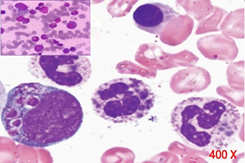
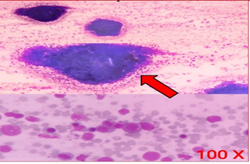
figure 3: hypercellular fragments with cytoplasmic granules in all myeloid lineage
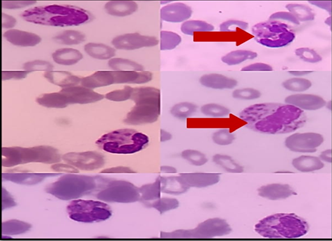
Microscopic examination of the hair shaft demonstrated the classical pattern of disease in the form of regularly distributed deposits of clumped melanin granules. (figure 4).

figure 4: a light microscope examination of hair shows abnormal clumps of melanin
Furthermore, luckily the diagnosis was supported further by homozygous mutation in lyst gene upon molecular analysis (table 2).
table 2: diagnostic criteria for hlh (accelerated phase of chediak higashi syndrome)
|
Criteria for accelerated phase ( hlh ) |
Patient findings |
|
1. Fever |
Fever ( 101f ) |
|
2. Splenomegaly |
Splenomegaly |
|
3. Bi / pancytopenia in the
peripheral film Hb<9.0g/dl Platelets<100× 109/l Neutrophils<1×109/l |
Hb: 7.6 g/dl Platelets: 20×109/l |
|
4. Hypertriglyceridemia >
3mmol/l Hypofibrinogenemia <1.5g/l |
Triglycerides : 3.33mmol/l Fibrinogen: 0.8g/l |
|
5. Ferritin > 500ng/ml |
Ferritin : 579ng/ml |
|
6. Hemophagocytosis |
|
|
7. Loss of nk cell activity |
Absent nk cell activity (recurrent infection) |
|
8. Sil-2r>2400u/ml |
|
Therefore,the patient was started on broad-spectrum antibiotics along with dexamethasone and cyclosporine. Later on, patient was refered to the bone marrow center where a successful hematopoeitic stem cell transplant was done upon remission of the accelerated phase.
After 6 months of transplant, the girl is being followed up by a multidisciplinary team and is doing well so far. Immunological and hematological defects have resolved with absence of any neurological manifestation.
Discussion
Beguez-cesar was the first physician to describe chediak higashi syndrome in 1943, followed by chediak and higashi who described the maldistribution of myeloperoxidases in the neutrophilic granules of affected patients6. The mean age of onset is 6 years; however, most patients die before the age of 107.
The lyst or chs 1 gene located on the long arm of chromosome 1(lq42-43) is associated with the development of chediak higashi syndrome. This gene is involved in the lysosomal trafficking transport of cytoplasmic granules. The 40 mutations consisting of nonsense, missense with deletion, and insertion4 disrupt protein synthesis, secretory functions, and storage of lymphocytic granules (figure 5)8.
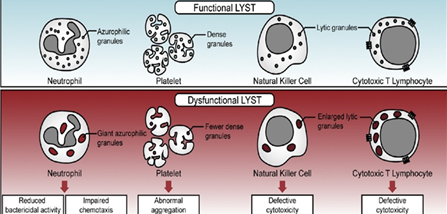
figure 5: an image showing pathophysiology of chediak higashi syndome9.
Figure obtained from: sharma p, nicoli er, serra-vinardell j, morimoto m, toro c, malicdan mc, introne wj. Chediak-higashi syndrome: a review of the past, present, and future. Drug discovery today: disease models. 2020 jun 1;31:31-6.
Clinically this syndrome is identified by silvery grey hair, hypopigmented skin, bleeding diathesis, and recurrent respiratory and gastrointestinal infections10, most commonly due to staphylococcus aureus and beta-hemolytic streptococcus11.
The majority of the patients likewise go through an “accelerated phase,” characterized by lymphohistiocytic infiltration of multiple organs precipitated particularly by epstein-barr virus (ebv) resulting in the development of anemia, bleeding tendency, hepatosplenomegaly, and life-threatening infections.
The presence of peroxidase-positive cytoplasmic granules mainly in neutrophils is the hallmark of chediak higashi syndrome3. Other differentials include the griscelli syndrome, hermansky pudlak syndrome, and elejalde syndrome.
The patients presenting in the accelerated phase must be brought into remission by using combination therapy consisting of dexamethasone, cyclosporine, and etoposide (according to hlh 2004 protocol), before performing the definitive treatment of hematopoietic stem cell transplantation2. Though this treatment cures the hematologic and immunologic defects, but this therapy does not prevent the progressive neurological deficit frequently observed during long-term follow-up5.
Conclusion
Chediak higashi syndrome is a unique disease with a diverse spectrum of clinical presentation and investigations. The pattern that we saw throughout the literature on this syndrome is a high mortality rate due to late diagnosis of this condition. So, we wanted to add on a survival story of a now four-year-old child from a developing country living a healthy, happy life due to prompt diagnosis and management. We hope this case of ours serves as a ray of hope for all those out there who have chediak higashi syndrome.
Patient consent
Consent was taken from the father of the patient for this case report.
References
1. kaplan j, de domenico i, ward dm. Chediak-higashi syndrome. Current opinion in hematology 2008;15(1):22-29.
2. gopaal n, sharma jn, agrawal v, lora ss, jadoun ls. Chediak-higashi syndrome with epstein-barr virus triggered hemophagocytic lymphohistiocytosis: a case report. Cureus 2020;12(11):e11467.
3. shiflett sl, kaplan j, ward dm. Chediak-higashi syndrome: a rare disorder of lysosomes and lysosome-related organelles. Pigment cell res 2002;15(4):251-257.
4. bouatay a, hizem s, tej a, moatamri w, boughamoura l, kortas m. Chediak-higashi syndrome presented as accelerated phase: case report and review of the literature. Ind j hematol blood transfus. 2014;30:223-226.
5. chandrakasan s, filipovich ah. Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. J pediatrics 2013;163(5):1253-1259.
6. maaloul i, talmoudi j, chabchoub i, et al. Chediak-higashi syndrome presenting in accelerated phase: a case report and literature review. Hematology/oncology and stem cell therapy. 2016;9(2):71-75.
7. Imran t, zafar l, rehan m, nasir a, tariq pa, batool i. Chediak-higashi syndrome presenting in accelerated phase. J coll physicians surg pak 2012;22(8):539-541.
8. bowman sl, bi‐karchin j, le l, marks ms. The road to lysosome‐related organelles: insights from hermansky‐pudlak syndrome and other rare diseases. Traffic 2019;20(6):404-435.
9. sharma p, nicoli er, serra-vinardell j, et al. Chediak-higashi syndrome: a review of the past, present, and future. Drug discov today dis models 2020;31:31-36.
10. patne sc, kumar s, bagri nk, kumar a, shukla j. Chediak-higashi syndrome: a case report. Indian j hematol blood transfus 2013;29(2):80-83.
11. introne w, boissy re, gahl wa. Clinical, molecular, and cell biological aspects of chediak-higashi syndrome. Mol genet metab 1999;68(2):283-303.