A Rare Case of Berardinelli-Seip Syndrome in a Four Month Old Child
ABSTRACT
Berardinelli-seip
congenital lipodystrophy [BSCL] or congenital generalized lipodystrophy [CGL]
is an extremely rare autosomal recessive disorder. It is one of the four
subgroups of lipodystrophy syndrome characterized by varying degrees of loss of
adipose mass in the body. The reported clinical presentation of this syndrome
include muscular hypertrophy, gigantism, hepatomegaly, impaired glucose
tolerance,acanthosis Nigricans, intellectual impairment, phlebomegaly,
hypertriglyceridemia, bone cysts and cardiomyopathy. Diabetes mellitus,
hypertriglyceridemia, and hepatic steatosis ordinarily develop in these
patients, and most girls suffer from menstrual abnormalities. We report a case
of an 8-year-old female child presented multiple times in the hospital with
complaints of failure to thrive and a marasmic look. She has lipoatrophy
affecting the face,limbs and trunk, acromegaloid features, hepatomegaly,
hypertriglyceridemia, phlebomegaly and hirsutism. Such patients need to be
identified and monitored for complications like diabetes mellitus and
cardiomyopathies.
Keywords:
Berardinelli-seip congenital lipodystrophy; Hypertriglyceridemia; Phlebomegaly;
Hepatomegaly
INTRODUCTION
Berardinelli-seip
syndrome is a rare genetic autosomal recessive disorder1, 1first described in 1954 by Berardinelli and
Seip2, 2affecting approximately 1 in
10 millions births worldwide. The earliest genetically confirmed case of
Pakistani origin is reported in 2013 in a consanguineous family.
On
the basis of alterations, 4 types of BSCL have been recognized as BSCL 1-4,
with their etiology linked to mutations in four distinct genes: AGPAT2, BSCL2, CAV1
and CAVIN1, respectively4. The common feature in all of these is loss of
adipose mass, accompanied by metabolic abnormalities, muscular hypertrophy and
distinct physical features. This syndrome is associated with severe insulin
resistance, dyslipidemia, and high risk of developing type 2 diabetes mellitus,
cardiovascular disease, and other comorbidities. The diagnostic criteria has
been described in literature, which include major and minor criteria. The major
criteria include lipoatrophy, acromegaloid features, hepatomegaly, hypertriglyceridemia
and insulin resistance. Minor criteria include hypertrophic cardiomyopathy,
psychomotor retardation, hirsutism, bone cysts, precocious puberty especially
in females, and splenomegaly. The diagnosis of BSCL is suggested if three major
or two major plus two minor criteria is fulfilled or a specific gene mutation
is identified. We report a case of an 8-year- old female child, product of
consanguineous marriage that fulfilled the clinical criteria of BSCL.
CASE REPORT
PRESENTATION
A 4-month-old female
child presented at the pediatric department of POF hospital, Wah Cantt, in September 2016, with complaints of
failure to thrive, a marasmic look and abdominal distention for 1 month. Her
parents are first cousins, residents of Busti, Wah Cantt. Birth history was
unremarkable. She has 2 siblings with normal development. On examination, her
ofc was 39 cm[below 3rd
centile], and her weight was 5.2 kg[below 5th centile], and length was 63cm
[above 25 th centile] with a BMI of 13.11kg/m2. She had a muscular build
with generalized loss of fat, mainly from buttock, face,
legs and arms; thick hands and feet; hirsutism; and prognathism. She also had distended abdomen,
firm hepatomegaly without
any signs of cirrhosis
or portal hypertension (Figure 1).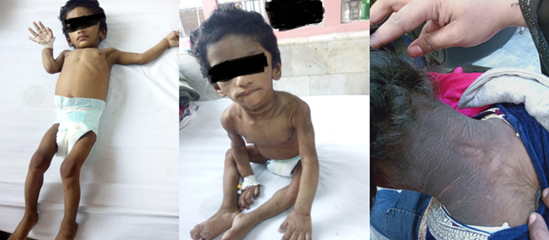
Figure 1:
Loss of subcutaneous fat, protuberant
abdomen, phlebomegaly and acanthosis nigricans
Chest examination was normal. There was hypertriglyceridemia
[655 mg/dl], raised ALT [122 IU/L] and ALP [322 IU/L].
CBC, Thyroid function
test, ferritin, vitamin
D, renal function
tests, and serum electrolytes were normal. Echocardiography was done
which was also normal.
Ultrasound abdomen showed hepatomegaly.
Initially, fasting and random sugar was in the normal range but increased later
on. Since the clinical criteria was met, the patient was diagnosed and treated as a case of BSCL,
however it was not confirmed due to unavailability of genetic testing. The patient was managed conservatively with
a low fat diet and increased physical activity along with close monitoring of
hypertriglyceridemia and blood sugar levels.
During
her course of illness, she developed acanthosis nigricans and her fasting blood
sugar level rose above 200 mg/dl. Her HBA1C was 9.3% for which she was
started on oral antidiabetics i.e metformin. Leptin was not
offered due to non-availability.
It was rare for patients with
Berardinelli-seip syndrome to develop diabetes during the first decade of life as our patient developed. Currently, the patient
is on followup in our department
and her blood sugars are well controlled on Metformin and she did not develop
any complications. [Parental consent was obtained for publishing the data with
photographs].
DISCUSSION
BSCL is usually diagnosed at birth or soon after birth. Because
of the absence of functional adipocytes, lipids, are stored in other tissues,
including muscle and liver. It is a rare disease, with a prevalence of approximately 0.96 cases/million and 500 cases
described in literature with fewer than 10
cases in Pakistan to date, with greater frequency reported in some ethnic
groups, mainly in Latin Americans
and Arabians [individuals of Portuguese and Norwegian ancestry]. It was first described in 1954 by Berardinelli in a 2-year-old
boy and later by Seip in three patients. BSCL etiology involves genetic
variations in four different genes: AGPAT2, BSCL2, CAV1 and CAVIN1. The four
different biochemical subtypes of the disease are distinguished depending on
which gene is mutated. The defect in BSCL is in the 1-acylglycerol-3-phosphate-
O-acyltransferase-2 [AGPAT2] gene on chromosome 9q34 in the type 1 variant and
in the BSCL2 gene on chromosome 11q13 in the type 2 variant.
Affected individuals develop insulin resistance [T2DM] and
approximately 25-35% develop diabetes between the ages of 15 and 20 years.
Hepatomegaly secondary to hepatic steatosis and skeletal muscle hypertrophy
occur in all affected individuals. Hypertrophic cardiomyopathy is reported in 20%-25% of affected individuals and is a significant cause
of morbidity from cardiac
failure and early mortality. Our patient developed firm hepatomegaly,
generalized lipodystrophy and hypertriglyceridemia. Clinically 4 major and 2
minor criteria for diagnosis were met by our patient. Our patient developed
diabetes in the first decade which is rare3
and usually developed
in the second decade of life.
Diagnosis of the disease is a
challenging task. For a definite diagnosis, genetic studies must be carried out. To date, no definitive treatment has been approved for BSCL. The management plan of these patients revolves around the
clinical manifestations and basic pathophysiology i.e. loss of metabolically
active adipose tissue and leptin deficiency which were the primary causes of
metabolic complications. A leptin analogue called
Metreleptin has been approved by United States
Food and Drug Authority (FDA) for use in BSCL patients5. Management in our patient included restriction of total fat
intake to 20-30% of dietary caloric intake, enhanced physical activity and oral
Antidiabetic i.e Metformin6-9.
CONCLUSION
This case report
highlights the clinical
presentation, diagnostic journey,
and management challenges
associated with this rare syndrome. There is a need for increasing awareness about this condition
among physicians so that early diagnosis and management with a multidisciplinary approach can be carried
out to prevent metabolic and systemic complications. Genetic counseling plays an integral role in the management of
this disease. Continued research and patient
follow-up are essential
to improve therapeutic strategies and outcomes
for individuals affected by
this complex syndrome.
Patient consent:
Written
consent was obtained
from the patient's father to publish
this case.
References
1. Handelsman Y, Oral EA, Bloomgarden ZT, et al. The clinical approach to the detection of
lipodystrophy-An AACE consensus statement. Endocr Pract 2013;19:107-116.
2. Lima JG, Nobrega LH, Lima NN, et al. Causes of death
in patients with Berardinelli-Seip congenital generalized lipodystrophy. Plos One 2018.13(6).
3. Akinci B, Onay H, Demir T, et al. Natural history
of congenital generalized lipodystrophy: A nationwide study from turkey.
J Clin Endocrinol Metab 2016;101(7):2759-2767.
4. Craveiro Sarmento
AS, Ferreira LC, Lima JG, et
al. The worldwide mutational landscape of Berardinelli-Seip congenital
lipodystrophy. Mutat Res Rev Mutat Res 2019;781:30-52.
5. Van Maldergem
L. Berardinelli-Seip Congenital Lipodystrophy. Gene Reviews 1993.
6. Cheema HA, Malik HS, Waheed N, Mushtaq I, Fayyaz Z, Anjum MN. Berardinelli-Seip
Congenital Generalised Lipodystrophy. J Coll Physicians Surg Pak
2018;28(5):406-408.
7. Gomes KB, Pardini VC, Fernandes AP. Clinical and molecular aspects
of Berardinelli-Seip
Congenital Lipodystrophy (BSCL). Clin Chim Acta 2009;402:1-6.
8. Mork NJ. Rajka G, Halse J. Treatment of acanthosis nigricans
with etretinate (Tigason)
in a patient with Lawrence-Seip syndrome (generalized lipodystrophy).
Acta Derm Venereol 1986;66(2):173-174.
9. Ashraf S, Masood S, Naz F, Rashid J. Berardinelli Seip Syndrome: A rare case report. J Pak
Med Assoc 2022;72(5):969-971.