Acromegaly due to Pituitary Microadenoma Complicated by Cerebrovascular Accident - Case Report
Abstract
Acromegaly is a rare, chronic endocrine disorder characterized by excessive
production of growth hormone and Insulin-like Growth Factor -1 (IGF-1), which
can lead to a range of serious multisystem complications. Due to its gradual
onset and the delays often associated with diagnosis, many patients present
with advanced complications, with cardiovascular issues-such as acute ischemic
stroke-being particularly prevalent. These complications not only elevate
mortality rates but also significantly diminish quality of life. Early and
precise diagnosis, along with effective biochemical management, are crucial for
alleviating these systemic complications and improving patient outcomes. This
is particularly vital as active acromegaly, marked by high levels of growth
hormone and IGF-1, is linked to substantial risk factors like insulin
resistance, systemic hypertension and dyslipidemia. We describe the case of a
36-year-old male who, following an acute ischemic stroke, was diagnosed with
acromegaly due to a pituitary microadenoma and achieved effective biochemical
control through targeted medical treatment.
Keywords: Acromegaly;
Systemic hypertension; Dyslipidemia; Insulin resistance; Acute ischemic stroke.
The overproduction of growth hormone and IGF-1 in acromegaly leads to a wide range of systemic complications, significantly increasing the risk of mortality and diminishing the quality of life for patients4. As a result of the gradual onset of the condition and frequent delays in diagnosis, over 90% of patients already experience complications at the time of diagnosis, with an average delay of 5.5 years. Unfortunately, a longer delay is associated with a worse overall prognosis5. The most prevalent complications are linked to the cardiovascular system, including left ventricular hypertrophy, ischemic heart disease, arrhythmias, cardiomyopathy and abnormalities in heart valves like aortic and mitral regurgitation, as well as cerebrovascular accidents (CVAs)6. Acute ischemic stroke is among the most common cardiovascular complications, with 4.5% of acromegaly patients initially presenting with a CVA7. The causes of stroke in these patients are attributed to both local and systemic factors8. Locally, pituitary adenomas play a role, while excess growth hormone promotes arteriosclerosis and disrupts arterial collagen metabolism, leading to the formation of cerebral aneurysms. These aneurysms can become a site for thrombosis, resulting in ischemic strokes. Additionally, treatments for pituitary adenomas, such as transsphenoidal surgery and radiotherapy, can further increase the risk of stroke. In cases involving growth hormone and prolactin- secreting adenomas, prolactin enhances platelet aggregation, further increasing the likelihood of stroke8.
The overproduction of growth hormone and IGF-1 also contributes to metabolic complications such as insulin resistance, Type 2 diabetes mellitus, systemic hypertension, dyslipidemia, oxidative stress, endothelial damage and a hypercoagulable state, all of which significantly increase the risk of acute ischemic stroke8. Notably, stroke incidence is elevated in acromegaly patients who suffer from systemic hypertension. Moreover, excess growth hormone and IGF-1 are linked to a hypo fibrinolytic and hypercoagulable state, further exacerbating the stroke risk. Consequently, active acromegaly characterized by elevated growth hormone and IGF-1 levels results in multiple systemic risk factors, intensifying the likelihood of stroke8.
Case Presentation
A
36-year-old male presented with chief complaints of difficulty in using his
left upper and lower limbs, including difficulty in gripping objects and
difficulty in walking or standing up from a squatting position upon waking in
the early morning. He also had deviation of angle of his mouth to the right and
slurred speech. He had no history of headaches or blurred vision and did not
report any other symptoms. He had no known history of diabetes, hypertension or
coronary artery disease. He was a chronic smoker and alcoholic for five years
but has been abstinent for the past three years. On examination, he exhibited a
prominent forehead, a large fleshy nose, prognathism, acral enlargement and
acanthosis. His blood pressure was 110/70 mmHg. There was decreased muscle tone
in the left upper and lower limbs, with power of ⅗ and an extensor plantar
reflex in the left lower limb. A non-contrast CT (Computed Tomography) scan of
the brain revealed an acute infarct in the right middle cerebral artery (MCA)
territory. X- ray foot of the patient showed increased heel pad thickness,
suggestive of acromegaly (Figure 1). Routine blood tests show results
within the normal range (Table 1).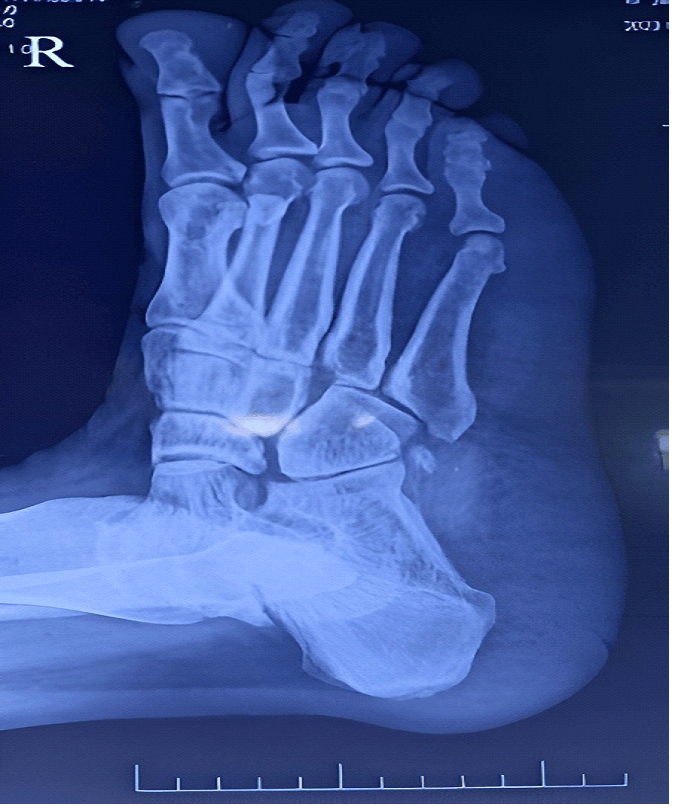
Figure 1: X-ray foot of the patient showing increased heel
pad thickness suggestive of acromegaly
Table 1: Complete Hemogram, Renal Function Test, Liver
Function Test, Serum Electrolytes, Calcium, Phosphorus, Magnesium, Pt, Inr,
Aptt
|
Parameter |
Observed value |
Reference value |
|
Hemoglobin |
12.8g/dl |
14-18g/dL |
|
Hematocrit |
38% |
42-50% |
|
White Cell Count |
7000/μL |
4000-11000/μL |
|
Platelet |
2.52 x 109L |
1.5-4.5 x109L |
|
Urea |
15 mg/dL |
8-20 mg/dL |
|
Creatinine |
0.7 mg/dL |
0.7-1.3mg/dL |
|
Total Bilirubin |
0.7 mg/dL |
0.3–1.0 mg/dL |
|
Direct Bilirubin |
0.2 mg/dL |
0.1–0.3 mg/dL |
|
SGOT(Serum Glutamic-Oxaloacetic Transaminase |
19 IU/L |
10–40 U/L |
|
SGPT( Serum Glutamic-Pyruvic Transaminase) |
16 IU/L |
10–40 U/L |
|
ALP (Alkaline phosphatase) |
84 IU/L |
35 – 130 IU/L |
|
Total protein |
7 g/dL |
6.5 – 8 g/dL |
|
Albumin |
4.4 g/dL |
3.5 – 5 g/dL |
|
Sodium |
143 mEq/L |
135–145 mEq/L |
|
Potassium |
4.0 mEq/L |
3.5–5.0 mEq/L |
|
Calcium |
9.2 mg/dL |
8.6–10.2 mg/dL |
|
Phosphorus |
3.2 mg/dL |
3.0–4.5 mg/dL |
|
Magnesium |
1.7 mg/dL |
1.6–2.6 mg/dL |
|
PT (Prothrombin time) |
16.4s |
11–13 seconds |
|
INR (International Normalised Ratio) |
1.28 |
<1.1 |
|
aPTT (activated Partial Thromboplastin Time) |
27.4 |
25–35 seconds |
MRI (Magnetic Resonance Imaging) brain revealed
encephalomalacia with adjacent gliosis in right frontotemporal region, T1
isointense and T2 hypo-intense lesion in the sella of size 9.1 × 8.8 × 8.6 cm.
On dynamic contrast, it shows hypo-enhancement and delayed homogeneous
enhancement suggestive of pituitary microadenoma (Figure
2).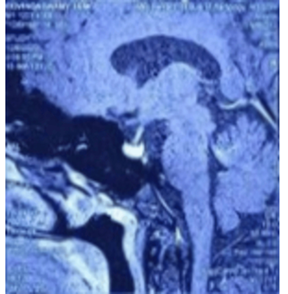
Figure 2: MRI brain with contrast showing homogeneous
enhancement of pituitary
We conducted a hormonal evaluation of the patient,
revealing elevated growth hormone levels and an age normalized IGF-1 that was
over 1.8 times the upper normal limit (Table
2)
Table 2: Fasting lipid profile, Hormonal profile of the patient
|
PARAMETER |
OBSERVED
VALUE |
REFERENCE
VALUE |
|
Total cholesterol |
149 mg/ dL |
Desirable - < 200
mg/dL |
|
Triglycerides |
79 mg/ dL |
Optimal - < 100 mg/ dL |
|
HDL (High Density
Lipoprotein) |
34 mg/ dL |
< 40 mg/ dL |
|
TSH (Thyroid Stimulating Hormone) |
1.0 microunits / mL |
0.5 – 4.0 microunits / mL |
|
Free T4 |
1.1 ng / dL |
0.8 – 1.8 ng /dL |
|
Growth Hormone |
5.7 ng / mL |
< 5 ng / mL |
|
IGF - 1 |
421 ng / mL |
83 – 232 ng /ml (
For 36 years) |
|
C - peptide |
2.0 ng / mL |
0.8 – 3.1 ng / mL |
|
Serum cortisol |
10.2 microgram / dL |
5 – 25 microgram /
dL |
|
ACTH (Adrenocorticotrophic hormone) |
53.8 pg / mL |
10 – 60 pg / mL |
|
FSH ( Follicle
Stimulating Hormone) |
8.25 milli IU/ mL |
1 – 7 milli IU / mL |
|
LH ( Luteinizing Hormone ) |
3.95 milli IU / mL |
2 – 9 milli IU / mL |
|
Serum prolactin |
19 ng/mL |
< 20 ng/mL |
|
Serum testosterone |
694.1 ng/dL |
291 – 1100 |
|
ng/dL |
In the growth hormone suppression test, after two
hours of the oral glucose tolerance test (OGTT), growth hormone level is 1.37ng
/ml and is not suppressed (Normally, should be < 0.4 ng/L for BMI < 25
kg/m2). As a result, he was diagnosed with acromegaly due to a pituitary
microadenoma. Echocardiogram showed a stable cardiac status and cerebrovascular
Doppler examination was normal. Hypercoagulability workup was conducted, shown in (Table
3).
Table 3: Hypercoagulability Workup
|
PARAMETER |
OBSERVED VALUE |
REFERENCE VALUE |
|
Anti beta – 2 glycoprotein antibody |
Negative |
- |
|
Anti – cardiolipin antibody |
Negative |
- |
|
Lupus anticoagulant |
Negative |
- |
|
Protein C |
122% |
70 – 140 % |
|
Protein S |
127% |
50 – 150 % |
|
Anti - thrombin lll |
113% |
70 – 140 % |
|
Factor V Leiden mutation |
Negative |
|
The patient was evaluated for additional
complications related to acromegaly. Both fasting and postprandial blood
glucose levels were within the normal range and the HbA1c (Glycated hemoglobin)
was 5.2%. During the hospital stay, the patient remained normotensive and the
fasting lipid profile was normal. Both ECG (Electrocardiogram) and
echocardiogram were normal. There was no evidence of visual field defects on
perimetry and the fundus examination was normal. A colonoscopy was performed,
revealing no evidence of colonic polyps or malignancy. An ultrasound of the
thyroid showed no thyroid nodules. There was no evidence of osteoarthritis or
signs of sleep disturbance. Dual-energy X-ray absorptiometry (DEXA) results
were normal.
In view of pituitary microadenoma and modest levels
of elevation of growth hormone and IGF - 1(< 2.5 times the upper limit of
normal), the patient was given a trial of dopamine agonist Cabergoline 0.25 mg
twice weekly and started on anti-platelets and regular physiotherapy done.
Patient was followed up at regular intervals.
After 6 months, IGF-1 level was 180 ng/ml and growth hormone suppression test was normal (Growth hormone level is 0.38 ng/ml after two hours of Oral Glucose Tolerance Test). Since the age normalized serum IGF-1 value and growth hormone suppression test was normal, the patient was maintained on medical management and advised regular follow up.
Discussion
Acromegaly is a rare and slowly progressing
disorder caused by chronic excess of growth hormone and IGF-1. This excess
leads to various physical changes and systemic complications, most commonly due
to a growth hormone-secreting pituitary adenoma1. Other rare causes include extra-pituitary growth hormone-secreting
tumors such as pancreatic islet cell tumors, lymphoma and central
GHRH-secreting tumors like pituitary hamartoma. Peripheral GHRH-secreting
tumors, including bronchial carcinoid, small cell lung carcinoma and medullary
thyroid carcinoma, can also be involved. Familial conditions like MEN1
(Multiple Endocrine Neoplasia), MEN4, FIPA (Familial Isolated Pituitary
Adenoma) and Carney complex, as well as the sporadic germline mosaic disorder
McCune-Albright disease, can increase the risk of pituitary hyperplasia and
neoplasia, potentially leading to acromegaly9. The
prevalence of acromegaly is about 5.9 per 100,000 people, with an incidence
rate of 0.38 cases per 100,000 person-years. Males are typically diagnosed
earlier than females, but females face a higher incidence and mortality risk,
lower IGF-I levels and experience a longer delay in diagnosis10,11.
Excess growth hormone and IGF-1 cause acral and somatic overgrowth. The most prevalent symptoms include acral enlargement such as prognathism/jaw enlargement oral changes like macroglossia, headaches, fatigue/tiredness, hyperhidrosis, snoring, skin changes such as oily skin, weight gain, arthralgia, maxillary widening causing teeth separation and acanthosis nigricans12. As the clinical features of acromegaly develop insidiously, diagnosis is often significantly delayed, with a mean diagnostic delay of 5.5 years.
Prolonged diagnostic delay is associated with increased mortality and morbidity5. Earlier recognition of the combination of clinical features is essential for establishing the diagnosis and initiating appropriate treatment. Most acromegaly patients present with complications due to diagnostic delays. Cardiovascular complications are the most common and include left ventricular hypertrophy, ischemic heart disease, arrhythmia, cardiomyopathy, diastolic and systolic dysfunction and valvular heart diseases such as aortic and mitral regurgitation, as well as cerebrovascular accidents (CVA). Other complications include metabolic issues like insulin resistance, hyperglycemia, dyslipidemia and neoplasms such as colonic polyps, colon carcinoma, breast carcinoma, thyroid carcinoma and endometrial and
The most frequent complications are left ventricular hypertrophy, hypercalciuria, endometrial polyps, fatty liver, diastolic dysfunction, thyroid nodules, hypertension, prediabetes, metabolic syndrome and digestive polyps12. Premature mortality in acromegaly patients occurs due to multiple systemic complications caused by excess growth hormone and IGF-1. The most common cause of death in the first decade is cardiovascular complications, while neoplasms remain the most common cause of death in the following two decades13. Men are generally younger than women at diagnosis and death, but women have increased mortality11,13. The systemic complications associated with acromegaly are as follows6,14.
Table 4: Systemic complications of acromegaly
|
SYSTEM INVOLVED |
COMPLICATIONS |
|
Cardiovascular system |
Left ventricular hypertrophy, Cardiomyopathy, Increased interventricular septum
thickness (eccentric hypertrophy), Diastolic and systolic Left ventricular
dysfunction, Ischemic heart disease, Cerebrovascular accident, |
|
|
Arrhythmia, Valvular heart disease - Mitral and Aortic regurgitation, Systemic hypertension, Endothelial dysfunction. |
|
Respiratory system |
Respiratory insufficiency, Sleep apnea syndrome
/ Obstructive sleep apnea. |
|
Metabolic complications |
Insulin resistance, Impaired fasting glucose, Impaired glucose
tolerance, Diabetes mellitus, Dyslipidemia -Reduced total cholesterol and increased triglycerides. |
|
Neoplasms |
Colonic polyp and colon carcinoma, Breast carcinoma, Thyroid
nodule and thyroid
carcinoma, Endometrial carcinoma. |
|
Musculoskeletal complications |
Osteoarthritis/Arthropathy,
Carpal Tunnel syndrome, Osteopenia, Vertebral fracture. |
|
Endocrine complications |
Multinodular thyroid goiter,
Thyrotoxicosis, Hypercalciuria, Hyperparathyroidism. |
Cardiovascular events are one of the leading causes of premature mortality in acromegaly patients15. Ischemic heart disease and cerebrovascular accident results in increased mortality and decreased quality of life in acromegaly patients.
Cardiovascular complications
Left ventricular hypertrophy (LVH) and
cardiomyopathy are connected to both the direct and indirect effects of IGF-16,15. Directly, IGF-1 promotes hypertrophy in cardiomyocytes, boosts
contractility by elevating intracellular calcium and has protective effects
against cell death. Indirectly, it lowers peripheral resistance by enhancing
nitric oxide release from the endothelium and increasing eicosanoids. For
diagnosing LVH, cardiac MRI is the preferred method as echocardiography can occasionally
provide an exaggerated assessment.
Diastolic dysfunction is commonly seen in patients with acromegaly, though it is usually mild and not clinically significant. Recent research shows that it generally does not progress to systolic dysfunction6. Furthermore, recent findings suggest that the incidence of myocardial infarction in acromegaly patients is comparable to that in the general population, with hypertension being a likely primary contributor6,15. Arrhythmias are more common due to fibrosis and IGF-1-induced changes in myocardial calcium channels, though they are typically clinically insignificant. Risk factors for valvular heart disease include the duration of acromegaly and elevated levels of growth hormone and IGF6,15. Systemic hypertension arises from increased plasma volume due to sodium and water retention, which is mediated by IGF-1 activation of the renin-angiotensin-aldosterone system6,15. Cerebrovascular accidents are one of the most important cardiovascular complications. About 4.5 % of the acromegaly patients presents initially with the CVA7.
Causes of cerebrovascular accidents in
acromegaly patients: Local
Extended exposure to growth hormone can lead to
arteriosclerosis, while changes in arterial collagen can result in cerebral
aneurysms. These aneurysms may serve as sites for thrombosis, potentially
causing ischemic stroke or they might rupture, leading to hemorrhagic stroke8. Elevated prolactin levels due to somatomammotropin adenomas can
enhance platelet aggregation, contributing to both arterial and venous
thrombosis8. Trans-sphenoidal surgery for pituitary
adenomas can induce cerebral vasospasm, which may lead to acute ischemic
stroke. Radiotherapy can exacerbate pre-existing cerebral angiopathy,
increasing the risk of acute ischemic stroke8.
Systemic
Acromegaly patients often have predisposing
risk factors for atherosclerosis, such as diabetes mellitus, hypertension and
dyslipidemia, which increases the risk for CVA8. Increased levels of circulating growth hormone are associated with a
hypo-fibrinolytic and hypercoagulable state, raising the risk of thrombosis. In
cases of active acromegaly, there is an elevation in fibrinogen, antithrombin
III, tissue plasminogen activator inhibitor 1 (PAI-1), while protein S and
plasma tissue factor pathway inhibitor levels are decreased.
Chronic systemic inflammation linked to acromegaly increases oxidative stress and endothelial dysfunction, further enhancing the risk of thrombosis. Elevated levels of growth hormone and IGF-1 activate signaling pathways such as STAT3(Signal transducer and activator of transcription 3, NF-κB(Nuclear factor kappa light chain enhancer of activated B cells), NLRP3 (Nucleotide binding domain, leucine rich containing family, pyrin domain containing-3)inflammasome and MAPK(Mitogen-activated protein kinase), leading to systemic inflammation and contributing to cardiovascular complications, including cerebrovascular accidents (CVA)8. Elevated IGF-1 levels are associated with endothelial dysfunction and cardiovascular disease (CVD). However, research on IGF-1 as an independent risk factor for CVD has produced mixed results and there is no clear evidence of a direct role of IGF-1 in atherosclerosis and CVD. The relationships might be influenced by how GH and IGF-1 affect underlying risk factors like hypertension, insulin resistance, inflammation and oxidative stress.
Diagnosis
In a patient with typical clinical signs and
symptoms, age normalized IGF-I > 1.3 times the upper limit of normal
confirms the diagnosis. In OGTT (Oral Glucose Tolerance Test), BMI (Body Mass
Index)-based GH nadir cutoffs can be considered for diagnosis, with < 0.4
µg/L for BMI < 25 kg/m2 and < 0.2 µg/L for BMI ≥ 25 kg/m2.
Treatment
Surgical resection of the pituitary adenoma is
the primary treatment for managing hyperpituitarism. Medical therapy is
recommended when surgery is not feasible and for patients with persistent
disease despite surgical resection of adenoma. The medical treatment involves
Somatostatin Receptor Ligands(SRL) such as octreotide, lanreotide and
pasireotide; a GH receptor antagonist like pegvisomant; and a dopamine agonist
like cabergoline. Cabergoline, a dopamine agonist, is recommended either as a
first-line treatment or as an adjunct to first-generation Stomatostatin
Receptor Ligands when IGF-1 levels are only mildly elevated, that is, less than
2.5 times the upper limit of normal. Radiotherapy can control tumor size but
may lead to cerebrovascular issues, hypopituitarism and cranial nerve damage8.
Achieving biochemical control by normalizing growth hormone and IGF-1 levels remains the strongest predictor of patient outcomes. It results in improved outcomes for complications like diabetes mellitus, obstructive sleep apnea (OSA), cardiovascular disease and vertebral fractures, but has the least effect on structural heart and joint changes11. To address elevated mortality in conditions associated with growth hormone (GH) and insulin-like growth factor I (IGF-I), achieving a serum GH level of less than 1 microgram per liter (µg/L) using radioimmunoassay (RIA) and normalizing serum IGF-I values for age is crucial. This target helps manage the condition and restore mortality rates to normal levels. Regular monitoring and adjustment of treatment are typically necessary to maintain these levels effectively. Normalizing IGF-1 is crucial to prevent comorbidities such as hypertension, cardiac hypertrophy, diabetes mellitus, glucose intolerance, sleep apnea and osteopathy. For patients with uncontrolled disease, aggressive management of these comorbidities is essential to prevent excess mortality. Once GH and/or IGF-1 levels are controlled, regular follow-up every six months is recommended.
Hypertension remains a significant contributor to cardiovascular mortality in acromegaly and may persist even with biochemical control. Management should adhere to general population guidelines. Acromegaly often causes insulin resistance, so diabetes management should follow general guidelines, with metformin as a first-line treatment. Pegvisomant can improve glucose metabolism in patients with inadequate response to first-line therapies8.
Dyslipidemia should be managed according to general population guidelines, taking into account the presence of other metabolic comorbidities. Acromegaly is also linked to an increased risk of colonic polyps and related conditions. Given recent advances in managing acromegaly and its comorbidities, cancer has become the leading cause of death8. IGF-1 levels correlate with colonic epithelium proliferation and the most common cancers in acromegaly are thyroid neoplasms and colon cancer. However, the rate of thyroid malignancies in acromegaly patients is similar to that in the general population8. Guidelines suggest performing at least one colonoscopy at diagnosis, with further surveillance based on initial findings and disease activity.
The follow-up recommendations include, blood pressure should be checked at diagnosis and every six months or with any change in antihypertensive treatment. An ECG and echocardiogram are recommended annually if abnormalities are detected. Colonoscopy should be performed every 10 years or more frequently if the initial results are abnormal, if IGF-1 levels remain elevated or if there is a family history of colorectal cancer. A dual-energy X-ray absorptiometry (DEXA) scan is advised every two years to assess bone density, while thoracic and lumbar spine X-rays should be conducted annually. Hormone levels, including total testosterone, sex hormone-binding globulin, prolactin, LH (Luteinizing hormone), FSH (Follicle-stimulating hormone), 17β -estradiol and serum free T4, should be checked annually. Fasting blood sugar and HbA1c (Glycated hemoglobin) levels should be monitored every six months and a quality-of-life assessment should be performed annually to evaluate the patient's overall well-being.
Conclusion
Human
Subjects
Consent
for treatment and open access publication was obtained or waived by all
participants in this study.
Conflicts
of Interest
In
compliance with the ICMJE uniform disclosure form, all authors declare the
following:
Payment/services
info:
All
authors have declared that no financial support was received from any
organization for the submitted work.
Financial
relationships
All
authors have declared that they have no financial relationships at present or
within the previous three years with any organizations that might have an
interest in the submitted work.
Other
relationships
All
authors have declared that there are no other relationships or activities that
could appear to have influenced the submitted work
References
3.
Lugo G, Pena L, Cordido F. Clinical manifestations and diagnosis of acromegaly. Int J Endocrinol 2012:540398.
4.
Kasuki L, Rocha PDS, Lamback EB, et
al. Determinants of morbidities and mortality in acromegaly. Arch Endocrinol
Metab 2019;63:630-637.
5.
Esposito D, Ragnarsson O, Johannsson G, Olsson DS. Prolonged diagnostic delay
in acromegaly is associated with increased morbidity and mortality.
Eur J Endocrinol 2020;182:523-531.
6.
Gadelha MR, Kasuki L, Lim DST, et.al. Systemic
Complications of Acromegaly and the Impact
of the Current Treatment
Landscape: An Update. Endocr Rev 2019;1:268-332.
7.
Petrossians
P, Daly AF, Natchev E, et al. Acromegaly
at diagnosis in 3173 patients from the Liège
Acromegaly Survey (LAS) Database.
Endocr Relat Cancer 2017, 24:505-518.
8.
Alnaaim SA, Al-Kuraishy HM, Zailaie
MM, et al. The potential link between acromegaly and risk of acute ischemic stroke in patients with pituitary
adenoma: a new perspective. Acta Neurol Belg 2024;124:755-766.
9.
Akirov A, Asa SL, Amer L, Shimon I, Ezzat S. The Clinicopathological Spectrum
of Acromegaly. J Clin Med 2019;13:8.
10. Dal J, Skov BG andersen M, et al. Sex differences in acromegaly at diagnosis: A nationwide cohort
study and meta-analysis of the
literature. Clin Endocrinol 2021;94:625-635.
11. Fleseriu M, Biller BMK, Freda PU, et
al. A Pituitary Society
update to acromegaly management guidelines. Pituitary 2021;24:1-13.
12. Slagboom TNA, van Bunderen CC, De Vries R, Bisschop
PH, Drent ML. Prevalence of clinical signs, symptoms
and comorbidities at diagnosis of acromegaly: a systematic review in accordance
with PRISMA guidelines. Pituitary
2023;26:319-332.
13. Ritvonen E, Löyttyniemi E, Jaatinen
P, et al. Mortality in acromegaly: a 20-year follow-up study. Endocr Relat Cancer 2016;23:469-80.