Adolescent’ Rhabdomyosarcoma of the Infra-Temporal Fossa: Case Report and Review of the Literature
Abstract
Rhabdomyosarcoma (RMS) is a malignant tumor of
striated muscle. It is the most common malignant mesenchymal tumor. The
prognosis of these tumors depends on the absence of metastasis at diagnosis,
the age of the child, and the location, volume and operability of the tumor.
The etiology of RMS is unknown, but certain syndromes such as neurofibromatosis
type 1, Li-Fraumeni syndrome, Beckwith-Wiedman syndrome and Costello syndrome
have been incriminated. Therapeutic management is based on surgical treatment
whenever possible, as well as postoperative chemotherapy/radiotherapy. In this
context, we present the case of a very advanced RMS of the infra-temporal fossa
in a 16-year-old child.
Key words: Rhabdomyosarcoma;
Infra-temporal fossa; Case report; Surgery
Introduction
Developed from primitive mesenchymal cells involved in
striated muscle differentiation, rhabdomyosarcoma (RMS) can occur anywhere in
the body. In terms of extra-cranial solid tumors in children, RMS is the third
most common after neuroblastoma and nephroblastoma1.
Rhabdomyosarcomas that occur in the head and neck
region, the most frequent site is the orbit, followed by the nasopharynx and
paranasal sinuses. The infratemporal fossa, on the other hand, is an extremely
rare site of such tumors. It is a highly malignant tumor, distinguished from
other sarcomas by its locoregional aggressiveness. Early diagnosis and
multidisciplinary management are therefore crucial2.
In this context, we report the case of an adolescent
who presented to our department with a symptomatology suggestive of
rhabdomyosarcoma of the infra-temporal fossa. Given its location, a diagnostic
biopsy was performed under general anesthesia before referring the patient for
chemotherapy/radiotherapy.
Case
report
We present the case of a 16-year-old adolescent, with
no particular medical history. Admitted to our ENT department for a right
parotid swelling, with no other associated signs, notably no dyspnea, dysphagia
or signs of facial paralysis. The patient's general condition remained stable. On
physical examination, the parotid mass is firm, fixed and sensitive to
palpation, with no inflammatory signs (Figure
1). Rhinoscopy shows a bulging of the nasopharynx. Endo-buccal examination
shows a bulging of the soft palate (Figure
2).
Cervical examination showed no abnormalities, notably
no palpable lymph nodes. 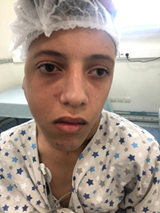
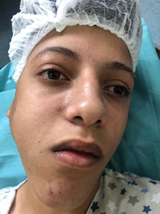
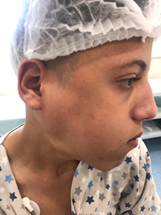
Figure 1: Image of the patient
showing the tumefaction opposite the parotid region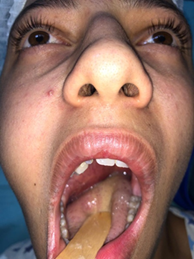
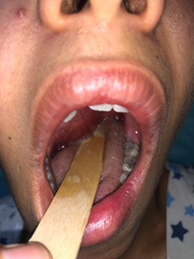
Figure 2: Image showing a bulging of
the soft palate
The patient then underwent MRI of the face, which
revealed a voluminous, largely necrotic tissue lesion in the right
infra-temporal fossa, measuring 94 mm in long axis, with irregular contours in
T1 hyposignal, discrete T2 hyper signal, diffusion hypersignal with low ADC,
intensely and heterogeneously enhancing after injection of gadolinium,
delineating areas of necrosis.
Superiorly and medially, it infiltrates the right parapharyngeal space
and the nasopharynx, partially filling its lumen. Superiorly and laterally, it
compresses the homolateral parotid gland and reaching the subcutaneous soft
tissue (Figure 3).
Inwardly and inferiorly, it infuses the hypopharynx
and the proximal part of the larynx, which remain permeable (Figure 5). Downwards and outwards, it
infiltrates the ascending branch of the mandible, with bone lysis (Figure 4). Anteriorly, it comes into
contact with the right maxillary sinus (Figure
6). Posteriorly, it fills the pre-stylial and retro-stylial space and
infiltrates the pre-vertebral muscles on the homolateral side. At the top, it
comes into contact with the floor of the orbit, with bone lysis of the anterior
and base of the skull, and infiltrates the homolateral temporal lobe (Figures 7 and 8). Below, it
infiltrates the right submandibular gland and the base of the tongue (Figure 5).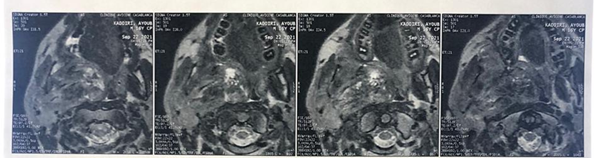
Figure 3: MRI on axial section
showing the tumor that infiltrates the right parotid, the right parapharyngeal
space and the nasopharynx, partially filling its lumen
Figure 4: MRI on coronal section
showing the tumor infiltrating the ascending branch of the mandible, with bone
lysis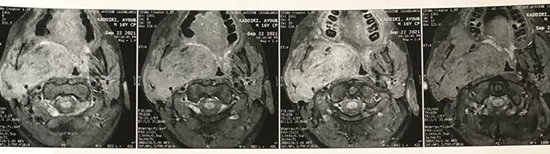
Figure 5: MRI on axial section
showing the infiltration of the base the tongue and the hypopharynx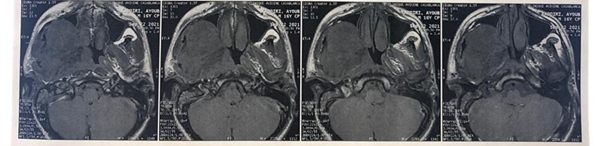
Figure 6: MRI on axial section
showing the infiltration of the right maxillary sinus.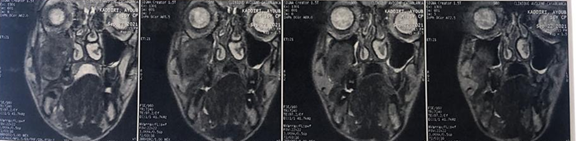
Figure 7: MRI on coronal section
showing the tumor that comes into contact the floor of the orbit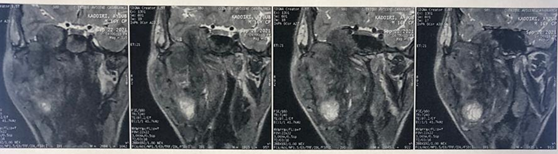
Figure 8: MRI on coronal section
showing the tumor infiltrating the base of the skull, and the homolateral temporal lobe
On this basis, the team decided to perform a biopsy
under general anesthesia for histopathological confirmation, followed by
chemotherapy/radiotherapy. The evolution was marked by a clear reduction in
tumor volume. The patient is still under close follow-up to detect any
recurrence or complication related to the tumor or radio-chemotherapy.
Discussion
Rhabdomyosarcoma (RMS) represents the most prevalent
mesenchymal tumor in children and adolescents. This is a malignant tumor of
unknown etiology with more or less marked striated muscle differentiation, of
mesenchymal origin. It can develop anywhere in the body, including sites where
striated muscle tissue does not normally exist. The most common sites are the
head and neck (40%). There are 350 new cases of rhabdomyosarcoma diagnosed
every year in the United States, with an annual incidence estimated at 8/10000001,2.
About two-thirds of RMS cases are diagnosed in
children under the age of six years, with two peaks in incidence under five and
in adolescents3.
Epidemiological data suggest that genetic factors play
an important role in the etiology of at least some of these sarcomas. A number
of specific genetic syndromes may be associated neurofibromatosis type 1,
Rubinstein-Tayebi syndrome, Wiedemann-Beckwith syndrome, Costello syndrome,
Noonan syndrome and Gorlin basic nevus syndrome. Among a series of 33 cases of
sporadic RMS, evidence of a germline p53 mutation was found in 3 of 13 children
under 3 years of age3-5.
This tumor is distinguished by rhabdomyoblasts,
slightly elongated cells with intracellular cross streaks and eosinophilic
cytoplasm6. Diagnosis relies to a
crucial extent on immunohistochemical analysis. RMS express Vimentin,
testifying to the conjunctival origin of cell proliferation, smooth muscle
actin (SMA) of striated muscle, and desmin, testifying to an intermediate
filament between smooth and skeletal muscles. Myo-D1 or Myf-3 are specifically
expressed in the nucleus of rhabdomyosarcoma cells in 80% of cases. Embryonal
rhabdomyosarcoma is confirmed by immunohistochemistry with positive markers
desmin, HHF35, possibly myoglobin and MyoD1; some cells may be cytokeratin
positive and PS100 positive7,8.
The most common molecular signature of ASMR involves
the chromosomal translocation t(2 ;3)(q35 ;q14)9.
In the case of embryonal rhabdomyosarcomas, differential diagnosis is often
made with other round cell tumors, such as neuroblastoma, lymphoma, PNET
(Peripheral Neuro Ectodermal Tumors), synovial-sarcoma or rhabdoid tumors10.
The primary site of rhabdomyosarcoma has long been
recognized as a key prognostic factor. It is also an important element to
consider in the therapeutic strategy for RMS, as it determines the quality of
the local procedure, with the existence of microscopic or macroscopic residue
to a greater or lesser extent. Orbit, Non-para meningeal head and neck, Para
meningeal head and neck, genitourinary organs and others (intra-thoracic,
intra-abdominal-pelvic, walls and perineum) are all potential sites of localization11,12.
Para-meningeal sites, involve the base of the skull
(nasopharynx, sinuses, middle ear, infra-temporal fossa and pterygopalatine),
with a propensity to erode adjacent bony structures and infiltrate intracranial
structures by contiguity13.
The American "IRS" system (Intergroup RMS
Study, United States) has developed a classification that takes into account
tumor operability. Group 1 or Localized disease, microscopic resection,
confined to the muscle or organ of origin without lymph node invasion. Group 2
where macroscopic resection is total but microscopic residue, regional disease
(extending beyond the muscle or organ of origin), completely resected or with
lymph node involvement. Group 3 Incomplete resection with macroscopic residue
or simple biopsy. Group 4 distant metastases at diagnosis10.
Concerning which imaging modality should be used when
a mass lesion of the infratemporal fossa is suspected, Levine et al reported a
representative case series in which malignancies that involved the
infratemporal fossa were better defined by axial MRI than by CT imaging14. (18).
It is important to consider at the time of diagnosis
both the possibility of a cure after well-managed treatment, and the acute and
late toxicities of the therapies used. Therapeutic management of these tumors
therefore requires a multidisciplinary approach involving the oncologist,
surgeon, radiotherapist, radiologist and pathologist3.
The surgical approach not only allows the biopsy
required for diagnosis, but also for complete primary removal of the tumour in
sites where extended removal would not compromise functional and/or aesthetic
prognosis, such as the limbs and trunk, or for a secondary removal leading to
better local control after reduction of tumour volume by chemotherapy and/or
radiotherapy. In head-neck cases, surgery is often limited to diagnostic
surgical biopsy15,16.
Chemotherapy has transformed the prognosis of these
tumors. These drugs are always used in combination, depending on the protocol.
Drugs with proven efficacy include actinomycin D, cyclophosphamide,
vincristine, cisplatin, carboplatin, dacarbazine (DTIC) and doxorubicin. More
recently, ifosfamide and etoposide have been added to the therapeutic arsenal.
Duration and intensity of treatment vary according to initial prognosis and
response to therapy17.
Radiotherapy's aim is to achieve local control, or to
consolidate that achieved by chemotherapy.
Doses used in recent studies range from 40 to 45 Gy for microscopic
disease control, and 50 to 55 Gy in the case of macroscopic residue10.
In terms of prognosis, we classify initial sites into
two categories, Favorable (involvement of the orbit, non-paramedic head and
neck, genitourinary region and bladder-prostate, with overall survival rates of
around 80% respectively). And unfavorable (involvement of the para-meningeal
region, bladder and prostate, limbs and other sites, with an overall survival
of 60%)18.
Conclusion
A definitive diagnosis relies on pathological
examination plus immunohistochemical analysis. Despite improvements in
therapeutic management, the prognosis for children head and neck
rhabdomyosarcomas remains very poor, given the often initially advanced stage
of the disease, frequent inoperability in the case of basicranial extensions,
and high metastatic potential. The management of rhabdomyosarcomas is
multidisciplinary, involving multi-drug therapy, surgery and external
radiotherapy.
References
1. Zafad S, Harif M,
Benchekroun S. Les rhabdomyosarcome de l’enfant. Esp Médicale 2002;9(80):96-98
2. Pizzo PA, Poplack DG. Rhabdomyosarcoma and the
undifferentiated sarcoma. Principles & Practice of
Pediatric Oncology 5th Edition 2006:971-996.
3. D’Andon A, Vassal PG, Oberlin
O, Hartmann O.
Tumeurs mésenchymateuses malignes ou sarcomes des parties molles. Institut
Gustave Roussy 2004;1-14.
4. Miller RW, Rubinstein JH. Tumors in Rubinstein-Taybi
syndrome. Am J Med Genet 1995;56(1):112-115.
5. Goldberg NS, Collins FS. The hunt for the
neurofi-bromatosis gene. Arch Dermatol 1991;127:1705-1707.
6. Grufferman S, Schwartz AG, Ruymann FM, et al. Parents
use of cocaine and marijuana and increased risk of rhabdomyosarcoma in their
children. Cancer Causes Control 1993;4(3):217-224.
7. Galmiche
L. Prise en charge anatomo-pathologique des tumeurs pédiatriques. Revue
Francophone des Laboratoires 2017;2017(488):49-58.
8. Shouman T, El-Kest I, Zaza K, Ezzat M, William H,
Ezzat I. Rhabdomyosarcoma in Childhood: A retrospective analysis of 190 patients
treated at a single institution. J Egyptian Nat Cancer Inst 2005;17(2):67-75.
9. Turc-Carel C, Lizard-Nacol S, Justrabo E, Favrot M,
Philip T, Tabone E. Consistent chromosomal translocation in alveolar
rhabdomyosarcoma. Cancer Genet.Cytogenet 1986;19:361-362.
10. Kalifa C, Pein F, Lemerle
J, Oberlin O, Hartmann O. Cancer de
l’enfant. Lavoisier MSP 2008.
11. Rodary C, Flamant F, Rey
A, et al. A report of common
critéria in chilhood rhabdomyosarcoma. J
Clin Oncol 1987;6:324-325.
12. Bergeron
C, Ranchere-Vince D, Berard-Marec P. Actualités sur le rhabdomyosarcome chez
l'enfant. Bulletin de cancer 2002;89(1):108-112.
13. Benk V, Rodary C, Donaldson SS, et al. Parameningeal
rhabdomyosarcoma: Results of an international workshop. Int
J Radiat Oncol Biol Phys 1996;36:533-540.
14. Levine PA, Paling MR, Black WC, et al. MRI vs.
highresolution CT scanning: Evaluation of the anterior skull base. Otolaryngol
Head Neck Surg 1987;96:260-267.
15. Neville HL, Andrassy RJ, Link MP, et al. Preoperative
staging: prognostic factors and outcome for extremity rhabdomyosarcoma: A
preliminary report from the Intergroup Rhabdomyosarcoma Study 4(1991-1997). J
Pediatr Surg 2000;35(2):317-321.
16. Blatt J, Snyderman C, Wollman MR, et al. Delayed
resection in the management of non-orbital rhabdomyosarcoma of the head and
neck in childhood. Med Pediatr Oncol 1997;29:294-298.
17. Maurer HM, Moon T,
Donaldson M, et al. The
intergroup rhabdomyosarcoma study: A priminary report. Cancer
1977;40:2015-2026.
18. Treuner J, et al. EpSSG
RMS2005 Protocol. 2008.