Atypical Presentation of Congenital Cholesteatoma in an Adult Case
Abstract
Cholesteatoma congenita is a recognized anatomoclinical entity. Adult forms are rare, with only a handful of documented cases. Making the diagnosis of congenital cholesteatoma in adult patients still pose a challenge even among experienced otologists. The etiopathogenesis of this lesion is still unknown; however, when cholesteatoma develops in subjects with no history of ear inflammation, as in the case we report here, an embryological origin is strongly suspected. An acquired origin is assumed in patients with a history of inflammatory processes in the outer or middle ear, due to proliferation of the basal cell layer of the tympanic membrane epithelium.it can occur in atypical places and at atypical ages. Many authors prefer tympanomastoidectomy of the canal wall. But it can also be successfully treated by intact canal wall tympanomastoidectomy, with good hearing results. We report a rare case of congenital cholesteatoma in an adult patient.
Key words: congenital cholesteatoma; adult; atypical presentation; conductive hearing loss; mastoidectomy
Introduction
The
first definition of congenital cholesteatoma with criteria to distinguish it
from acquired cholesteatoma was proposed by derlacki and clemis1 in 1965 and completed by levenson et al.
In 19892. They proposed the
following criteria:
• the existence of a white mass on
the medial aspect of a normal eardrum1,2;
• a normal pars flaccida and pars
tensa1,2;
• absence of antecedent otorrhea or
perforation2;
• no history of myringotomy or
middle ear surgery2;
• exclusion of intratympanic or
giant cholesteatoma2;
• antecedent otitis media is no
longer an exclusion criterion2.
It may
be discovered incidentally by otoscopy. The typical appearance is that of a
whitish retrotympanic mass. Its location is variable, with, in order of
frequency, the anterosuperior mesotympanum (65%), the posterosuperior meso
tympanum (15%), the two upper quadrants (17%) and the anteroinferior
mesotympanum (2-5%)2.the
otoscopic appearance may also be strictly normal5.
It can also be diagnosed by aspiration of epidermal debris during paracentesis
for osm or suspected in the presence of conductive hearing loss or unilateral
seromucous otitis. The persistence of conductive hearing loss in a child after
placement of a tympanic should raise the possibility of congenital
cholesteatoma. Facial paralysis (9%)5
and rarely vertigo or tinnitus may complete the clinical picture. Many reports
of pediatric cholesteatomas exist but studies in adult population are
relatively less. We report one description in adults.
Case report
A 41-year-old lady presented to our ear, nose
and throat (ent) department with progressively worsening right hearing loss and
chronic right otalgia, patient without any particular pathological history. There
was no history of otitis media, ear trauma or otologic surgery. The history of
her illness goes back to 3 years with the installation of o-progressive right
hypoacusis with intermittent rotatory vertigo and intermittent headaches
without other associated signs in particular no facial palsy no tinnitus. The
evolution was marked by the appearance of intense and intermittent chronic
homolateral otalgia. In the examination, normal otoscopy (figure 1), there was no facial palsy, no spontaneous
nystagmus.
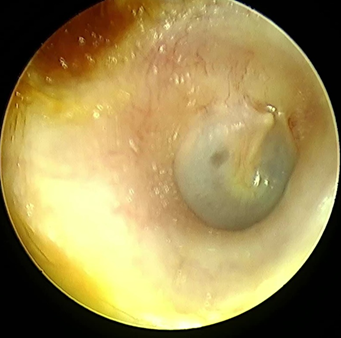
figure 1: otoscopy of the right ear
The
audiogram showed the conductive hearing loss on the right ear with a loss of 70
db and weber was lateralized to the right (figure 2). The tympanogram curve is flat on the right ear.
The video-head impulse test schowed a right lateral deficit (figure 3).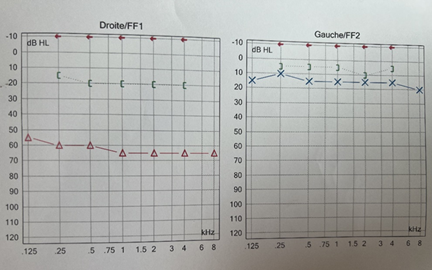
Figure
2: right ear showed a moderate conductive hearing loss with
air-bone gap of 45 dbh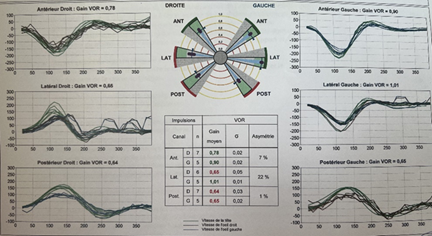
Figure 3: the video-head
impulse test schowed a right lateral deficit
On the ct scan of the temporal bone, the right
ear cavity had a soft tissue density, incomplete filling of the epitympanum and
mesotympanum, by well-defined, encapsulated tissue formations, one of which is
pedicled opposite the jacobson's nerve, thining of the tegmen tympani,
ossicular lysis, fistula between the upper semicircular canal and the tympanic
cavity and mastoid cell filling (figure
4).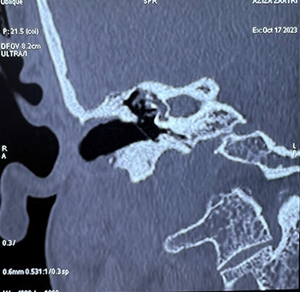
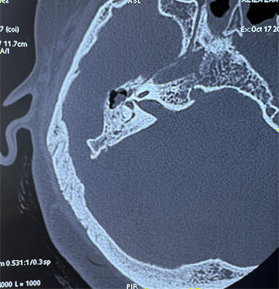
Figure 4: ct scan of the temporal bone in coronal and axial cuts: showing soft tissue density within the right middle ear cavity, pedicle tissue formation opposite the jacobson nerve, ossicular lysis, fistula between the upper semicircular canal and the tympanic cavity.
In the mr imaging of the temporal bone schowed
abnormal signal intensity within the right middle ear cavity which appeared
hypointense on t1-weighted, hyperintense on t2-weighted images with minimal
peripheral enhancement post-gadolinium, coincide with the diagnosis of
cholesteatoma. Mastoid cell filling are also filled in, with loss of the
annular appearance of the right semicircular canal (figure 5).
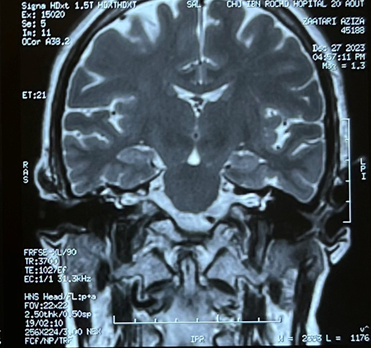
Figure 5: mr
imaging of the temporal bone schowed a loss of the annular appearance of the
right semicircular canal.
The patient underwent middle ear exploration, which confirmed the presence of a congenital cholesteatoma. The lesion was removed using a retroauricolar approach, following which a miringoplasty (underlay techinique) was performed using an autologous temporalis muscle fascia graft). Intra- or postoperative complication was seen in the patient. The diagnosis of cholesteatoma was confirmed histologically. The pure-tone average thresholds obtained at the 6th week of the operation were 10 db (air/bone thresholds respectively). Follow-up 3 months after surgery revealed no signs of recurrence.
Written informed consent was obtained from the patient for publication of this case report and accompanying images. All diagnostic and therapeutic procedures were performed with patient’s consent and respecting ethical principles of our institutional and national committee.
Discussion
Congenital cholesteatoma (cc) can appear at any age
from infancy to adulthood (mean 16.7 years of age) and it is caused by
congenital epithelial remains in the temporal bone6.
Its histological appearance is comparable to acquired cholesteatoma;
nevertheless, some characteristics of the patient, such as the absence of ear
illness or surgical history, support the diagnosis of cc7.
Otorrhea, the clinical sign of acquired cholesteatoma in children, is absent in congenital cholesteatoma5. Location is not a diagnostic criterion, but according to duclos et al5, potsic et al8 and zappia and wiet9, two clinical forms of cholesteatoma can be described cholesteatoma: localized anterosuperior encapsulated forms, asymptomatic and without hearing loss; diffuse forms with invasion of the posterosuperior quadrant or mesotympanum, frequently associated with ossicular erosion. Potsic et al point out that the finding of two different clinical forms is correlated with two population groups of different ages8. Encapsulated anterosuperior forms are more frequent in young children, whereas diffuse forms with invasion of the posterosuperior quadrant or mesotympanum are found in a wider population of children of varying ages8. Conductive hearing loss is an inevitable consequence of cc involving the posterior tympanum10. In the adults and elderly, the disease may involve an unusual localization and conductive hearing loss is the most common symptom in these patients.
Congenital cholesteatoma in adults is rare, but there
are a few descriptions:
·
Zappia and wiet report a
case of congenital cholesteatoma in a 30-year-old man.a case of posterosuperior
congenital cholesteatoma with erosion of the incus and head of the malleus has
been reported9.
·
Mc donald et al describe
five cases in patients aged 22 to 59 with conductive hypoacusis, intact
tympanic membrane, facial or dural dehiscence, labyrinthine fistulae or
multiple localizations10.
·
Mendoc¸a cruz et al
report a case of congenital cholesteatoma in a 26-year-old woman presenting
clinically with hypoacusis and associated facial palsy11.
·
Galli et al. Report a
case of congenital cholesteatoma in a 34-year-old man, revealed by
temporomandibular joint pain10.
·
Soderberg and dornhoffer
describe a case of congenital cholesteatoma in a 24-year-old woman with 55 db
of hearing loss, which developed progressively over several years12.
· With the exception of the case described by zappia et al11, adult forms are extensive and late-onset. In these cases, there is always labyrinthine involvement, facial paralysis and diffuse involvement of the middle ear or even the temporomandibular joint.
Most descriptions in adults are symptomatic of a diffuse, progressive form and the absence of otorrhea, perforation, myringotomy or middle-ear surgery, together with an intact eardrum, allow the diagnosis to be made, but the encapsulated form in the posterior-superior quadrant is not described.
More severe consequences such labyrinthine fistula, facial paralysis, meningitis, cranial abscess and even death could result from cc if it is discovered too late13.
Imaging is crucial to the diagnosing process, particularly when it comes to differentiating between lesions that impact the mastoid and the petrous apex. In magnetic resonance imaging (mri), cc have low signal on t1 and high signal on t2, they do not augment with contrast and they exhibit restricted diffusion on diffusion weighted imaging (dwi) sequences16. Computerized tomography (ct) displays expansile well circumscribed mass. Another function of dwi is to detect recurrent or residual disease, particularly in situations where the canal wall is preserved during surgery or when cartilage is utilized to reconstruct the tympanic membrane and direct otoscopic visualization is insufficient to determine the condition. As a result, dwi may prevent needless second opinion procedures17.
Congenital cholesteatoma is a condition primarily affecting children. 73% of patients who underwent surgery over a 24-year period were 15 years of age or younger, according to a sizable retrospective study14. The disease is generally in a more advanced stage when a patient is diagnosed, generally speaking15. When our case was diagnosed and operated on, he was 35 years old.
The unusual condition known as congenital cholesteatoma of the middle ear is caused by squamous epithelial cells in the tympanic cavity that were left over from embryonic development. Despite being innocuous at first, it worsens over time and can lead to potentially fatal complications. The prognosis for middle ear anatomy-related hearing loss is typically poor. These factors make early diagnosis and treatment of this illness crucial.
Recurrence rate and hearing improvement after canal wall down or wall up mastoidectomy have not been reported different in the literature in cases with cc18,19.
References
1. Derlacki el, clemis jd. Congenital
cholesteatoma of the middle ear and mastoid. Ann otol rhinol laryngol
1965;74(3):706-727.
2. Levenson mj, michaels l, parisier sc, et
al. Congenital cholesteatomas of the middle ear in children: origin and
management. Otolaryngol clin north am 1989;22(5):941-954.
3. Mahanta vr, uddin fj, mohan s, sharp jf.
Non-classical presentation of congenital cholesteatoma. Ann r coll surg engl
2007:89(2).
4. Jang ch, cho yb, kim yh, wang pc.
Congenital cholesteatoma associated with blue eardrum. In vivo
2009;23(1):163-166.
5. Duclos jy, darouzet v, portmann d,
portmann m, bebear jp, cholesteatomes congenitaux de l’oreille chez l’enfant
analyse clinique, e´volutive et the´ rapeutique d’une se´ rie de 34 cas : ann
otolaryngol chir cervicofac 1999;116(4):218-27.
6. Kojima h, miyazaki h, tanaka y, shiwa m,
honda y, moriyama h. Congenital middle ear cholesteatoma: experience in 48
cases. Nippon jibiinkoka gakkai kaiho 2003;106:856-865.
7. Weber pc, jr.adkins wy. Congenital
cholesteatomas in the tympanic membrane. Laryngoscope 1997;107(9):1181-1184.
8. Potsic wp, korman sb, samadi ds, wetmore
rf. Congenital cholesteatoma: 20 years’ experience at the children’s hospital
of philadelphia. Otolaryngol head neck surg 2002;6(4):409-414.
9. Zappia jj, wiet rj. Congenital
cholesteatoma. Arch otolaryngol head neck surg 1995;121:19-22.
10. Mc donald tj, cody dtr, ryan jr re.
Congenital cholesteatoma of the ear. Ann otol rhinol laryngol
1984;93(6):637-640.
11. Mendoc¸a cruz ol, alvares cruz filho n, maia
ra, miniti a, formigoni lg. Choleste´atome d’origine conge´nitale de l’apex et
de l’oreille moyenne. Rev laryngol 1982;103:227-229.
12. Soderberg kc, dornhoffer jl. Congenital
cholesteatoma of the middle ear, occurrence of an ‘‘open’’ lesion. Am j
otolaryngol 1998;19(1):37-40.
13. Sheehy jl, brackman de, graham md.
Complications of cholesteatoma: a report on 1024 cases. In: hattori bf, sade j,
abramson m, editors. Cholesteatoma: first international conference. Aesculapius
publishing; birmingham: alabama 1977;420-429.
14. Kojima h, tanaka y, shiwa m, sakurai y,
moriyama h. Congenital cholesteatoma clinical features and surgical results. Am
j otolaryngol head neck surg 2006;27(5):299-305.
15. Lim hw, yoon th, kang ws. Congenital
cholesteatoma: clinical features and growth patterns. Am j otolaryngol head
neck surg 2012;33(5):538-542.
16. Warren fm, bennett ml, wiggins rh, iii,
saltzman kl, blevins ks, shelton c, harnsberger hr. Congenital cholesteatoma of
the mastoid temporal bone. Laryngoscope 2007;117(8):1389-1395.
17. Schwartz km, lane ji, bolster bd, neff ba.
The utility of diffusion-weighted imaging for cholesteatoma evaluation. Am j
neuroradiol 2011;32(3):430-436.
18. Edfeldt l, kinnefors a, strömback k, köbler
s, rask-andersen h. Surgical treatment of paediatric cholesteatoma: long-term
follow up in comparison with adults. Int j ped otolaryngol
2012;76(8):1091-1097.
19. Park kh, park sn, chang kh, jung mk, yeo sw. Congenital middle ear cholesteatoma in children; retrospective review of 35 cases. J korean med sci. 2009;24(1):126-131.