Clinical Epidemiology and APOE Gene Etiology: An Update on Alzheimer’s Disease.
ABSTRACT
This
review discusses the discovery, epidemiology, and gene etiology of Alzheimer’s
disease (AD). It also highlights the Alzheimer’s gender disparity common
comorbid and modifiable risk factors. AD is a progressive neurodegenerative condition that impacts both daily
activities and social interactions. With the rise in life expectancy and
demographic aging, the global prevalence of AD is expected to increase further,
particularly in developing peoples, resulting in a significant burden of
disease. Its prevalence in the general population increases dramatically with
age; it affects approximately 80% of patients aged ≥ 75 years. AD that occurs
before the age of 65, known as early-onset AD (EOAD), is not as extensively
studied as late-onset AD (LOAD), even though EOAD often presents with a more
aggressive disease progression. AD is a complex and multifactorial disorder
influenced by genetic susceptibility and environmental factors over the
individual’s life. Sex-specific risk factors of
dementia for women, including pregnancy, menopause, and preeclampsia, account
for the rise of AD among women compared to men. Identifying modifier
genes has facilitated the control of APOE genes and their co-expressed genes.
Nevertheless, exploring new gene modifiers may enhance a better understanding
of intricate AD characteristics and hold potential therapeutic benefits for individuals
with AD.
Keywords: Alzheimer’s disease; Clinical epidemiology; Gene etiology;
APOE, gene modifiers; Functional enrichment analysis
BACKGROUND
Alzheimer's disease
(AD, MIM 104300) is the leading cause of dementia, characterized by a decline
in cognitive abilities, functioning, and behavior1, occurring
with approximately
50 million people currently living with dementia worldwide2. The aging
population is expected to cause this number to triple by 20503. This surge in cases poses a higher risk of disability,
increased burden of illness, and rising healthcare costs3. The prevalence and incidence of AD significantly rise
with age, reaching about 80% of patients aged 75 years or older. The incidence
rates increase from 2 per 1,000 individuals aged 65-74 to 37 per 1,000
individuals aged > 854. The majority of AD cases occur after reaching ≥
65 years and are commonly referred to as late-onset AD (LOAD), whereas cases
aged < 65 are infrequent (less than 5% of cases) and are classified as
early-onset AD (EOAD)5.
A precise
diagnosis is challenging for individuals experiencing cognitive dysfunction. The decisive
pathological characteristics found in the brain tissues of individuals with AD
include elevated levels of both amyloid-β (Aβ) forming extracellular senile
plaques and hyperphosphorylated tau (p-tau) accumulating intracellularly as
neurofibrillary tangles (NFTs)6. While AD is
typically indicated by Aβ and tau biomarkers, some cognitively normal
individuals who exhibit only these biomarkers do not progress to AD7,
highlighting the challenges in obtaining a pre-symptomatic diagnosis for those
individuals.
While there is
sufficient evidence on the prevalence of AD in Europe and North America, data
is scarce in South and Southeast Asia, Africa, the Middle East, Russia, Eastern
Europe, and Latin America8,9. Although there are no recognized
clinical trials for a cure, AD-modifiable risk factors and prevention,
including psychosocial interventions, education, and lifestyles, can help lower
the likelihood of AD or postpone its onset. This review discusses the discovery,
epidemiology, and gene etiology of AD, particularly APOE. It also highlights
AD’s gender disparity and specific comorbid and functional enrichment database
outcomes.
THE EARLY HISTORY OF AD
Earlier, in 1901, Alois
Alzheimer investigated a female patient admitted at Frankfurt Hospital-Germany,
who exhibited various behavioral and psychiatric symptoms, including paranoia,
delusions, hallucinations and impaired memory10. Later, the
National Institute of Neurological and Communicative Disorders and
Stroke-Alzheimer's Disease and Related Disorders Association (NINCDS-AD &
DA) outlined the most common criteria of the disease11. These
criteria for a probable diagnosis of Alzheimer's include dementia as determined
by the mini-mental state examination (MMSE),12 which allows a brief
quantitative measure of cognition status to be determined. It can be used to
measure cognitive decline, document cognitive changes over time and with
treatment, and as an effective tool in screening for elements of cognitive
impairment12.
EPIDEMIOLOGY
Incidence and Mortality Rates
The global number of individuals who
have dementia is estimated to reach 152 million by the middle of the century.
This increase is expected to be most significant in low- and middle-income
countries3.
The number of Alzheimer's patients aged 65 and above in The United States may
rise significantly from 5.8 million to 13.8 million by 2050 (2020 Alzheimer’s
disease facts and figures). The prevalence of AD has notably increased in
community-dwelling studies conducted in Japan and China over the past few
decades13,14.
Moreover, the age-specific global prevalence in women is 1.17 times higher than
in men. The age-standardized mortality rate for women is also greater,
indicating that a longer lifespan alone does not account for women's higher
prevalence15.
According to current estimates, there
are approximately 6.7 million individuals in the United States who are 65 years
and older and are currently living with Alzheimer's dementia16. However,
if there are no significant advancements in medical research to prevent, slow
down, or cure AD, this number could potentially increase to 13.8 million by
2060. In 2019, official death records documented 121,499 deaths caused by AD,
making it the sixth-leading cause of death in the United States16.
Furthermore,
numerous risk factors may contribute to the onset of AD and also manifest as
symptoms of AD at the same time, suggesting a potential reverse causality. Between 2020 and 2021,
COVID-19 emerged as one of the top ten leading causes of death, while AD held
the seventh position. Among Americans aged 65 and above, AD continues to be the
fifth-leading cause of death. Notably, from 2000 to 2019, fatalities resulting
from stroke, heart disease, and HIV declined, whereas reported deaths
attributed to Alzheimer's disease witnessed a significant increase of over 145%16.
Gender Disparities
Gender and age
disparities are seen in cognitive disturbances in various neurological and
psychiatric disorders. Multiple environmental, behavioral, and lifestyle
factors have been linked to AD, and the associations between several of these
risk factors and dementia differ based on sex and gender17. The number
of individuals affected by AD is increasingly rising at a greater rate in women
compared to men in the future. This trend is attributed to the increased
lifespan of women and biological factors4. The overall
risk of developing AD for individuals aged 65 is 21.2% for females and 11.6%
for males3,18. The expected survival duration for AD varies
from four to eight years across different studies. It is influenced by various
factors such as age at diagnosis, gender, behavioral characteristics,
involvement of the motor system, and co-existing medical conditions19.
Sex-specific risk factors of dementia for women, including pregnancy and
menopause, have demonstrated that a history of preeclampsia increases the risk
of mild cognitive impairment, vascular dementia, and AD20-23. Moreover,
early menopause before the age of 45 has been associated with an increased risk
of mild cognitive impairment and dementia.
It is still uncertain
if there is an association between low testosterone levels and the risk of
dementia in males24. However, sex steroids can have a
positive impact on brain function, and lower levels of these hormones may be
linked to poorer cognitive function in older men25,26. On the
other hand, previous studies have presented conflicting findings regarding the
relationship between testosterone levels and the risk of AD in older men27-29. A
meta-analysis study involving 5,251 older men and 240 cases of AD revealed a
significant association between low testosterone levels and an increased risk
of the disease (random relative risk = 1.48, P = 0.006)30.
Nevertheless, androgen deprivation therapy, a widely used treatment for
prostate cancer, has been linked to an increased risk of cognitive impairment
and dementia31.
CLINICAL CHARACTERISTICS OF AD
Age is commonly
recognized as a primary risk factor for AD and is utilized in two categories32,33. The two
primary subcategories of the disease encompass early-onset AD (EOAD) and
late-onset AD (LOAD). These classifications are assigned when individuals
typically exhibit symptoms, usually around 65 years old33. However,
it may occur earlier in cases where genetic mutations in familial Alzheimer's
disease (FAD) genes are involved. AD symptoms typically accompany numerous
cognitive impairments in various areas, including visuospatial, language, and
executive function34.
Late-Onset AD
Typically, the clinical
manifestation of AD is primarily marked by a significant decline in anterograde
episodic memory. This symptom is commonly seen alongside various cognitive
deficits like visuospatial abilities, language skills, and executive function34. The
presence of these specific AD features collectively results in an overall
cognitive deterioration, ultimately culminating in complete dependence and
mortality35. In the late stage of AD, magnetic resonance imaging may
observe ventricular enlargement and shrinkage of the brain. Various alterations
observed in the AD brain include neuronal loss in specific areas, intracellular
neurofibrillary tangles within the neurons of the cerebral cortex and
hippocampus, and neuritic plaques composed of amyloids that dystrophic
neurites, reactive astrocytes, and microglia could encircle36-38.
Early-Onset AD
Although the typical manifestation of
memory-predominant phenotypes overlaps between LOAD and EOAD cases, some EOAD
cases exhibit atypical patterns where episodic memory remains intact while
experiencing specific cortical symptoms associated with language, visuospatial
abilities, or executive function35. As the disease
progresses, a specific set of non-memory symptoms is observed in approximately
25% of cases with EOAD. These symptoms include apraxia, visual dysfunction,
fluent or non-fluent aphasia, executive dysfunction, or dyscalculia39-42. Significant
disparities in the onset age exist both within and across families, partly
attributed to genetic, environmental, or random factors41. While some
autosomal dominant cases develop first symptoms as early as their late 20s,
others develop the disease in their early 60s (prevalence increases with age)42. Finally43, reported that,
in families with PSEN1, PSEN2, or APP-caused AD mutations, the characteristics
of neuroticism and conscientiousness were linked to the time until symptoms
appeared, indicators of tau pathology in the cerebrospinal fluid (CSF), and the
progression of cognitive decline over time39.
MODIFIABLE RISK AND PROTECTIVE FACTORS
Multiple longitudinal investigations
have pinpointed a range of risk and protective factors associated with AD, some
of which may be addressed to lower the likelihood of AD or postpone its onset44-48. AD is
believed to begin decades before any noticeable clinical symptoms manifest. As
a result, addressing multiple risk factors in non-demented elderly individuals,
even the middle-aged population, could potentially help in either preventing or
postponing the onset of AD47. Efficient preventive efforts can potentially
hinder the progression of AD. Additional policies to promote education and
raise awareness about social or cognitive activities should be proposed to the
general public. In addition, maintaining healthy lifestyles and protecting
against air pollutants in the environment is crucial for preventing AD47 (Figure
1).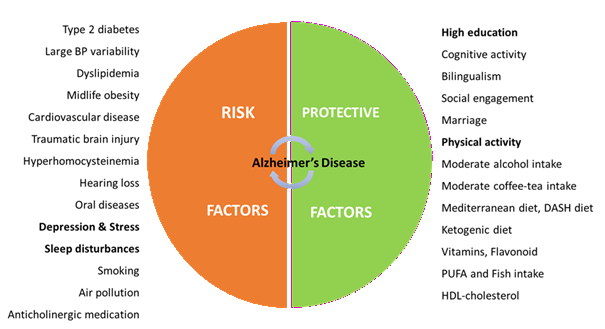
Figure 1. Modifiable risk and protective factors. Some factors appeared to be
risk factors and symptoms of AD, possibly due to the reverse causality, as
shown in bold.
Abbreviation: BP = blood pressure,
DASH = Dietary Approach to Stop Hypertension, MIND = Mediterranean-DASH diet
Intervention for Neurodegeneration Delay, PUFA = polyunsaturated fatty acid,
HDL-cholesterol = high-density lipoprotein cholestero47.
GENETIC
ETIOLOGY
The genes implicated in
early-onset forms of AD, which occur below 65 years of age, are the APP gene
located on chromosome 21q21 (MIM 104760); PSEN1 located on chromosome 14q24.3
(MIM 104311) and PSEN2 on chromosome 1q31-q42 (MIM 600759). Missense mutations
within the PSEN1 gene account for 18-50% of AD's early-onset autosomal dominant
forms49. Mutations within the PSEN1 gene lead to an aggressive
form of the disease, with an onset age between 30 and 50 years, which is not
influenced by the APOE genotype. However, a polymorphism found within intron 8
of the PSEN1 gene was associated with developing the late-onset form of AD50,51. PSEN1 and
PSEN2 account for less than 5% of AD cases51-54.
Regarding
the APP (MIM 104760), Glenner and Wong55 previously isolated a protein from the twisted beta-pleated sheet
fibrils found in cerebrovascular amyloidoses and amyloid plaques associated
with AD (MIM 104300)56.
determined in a comprehensive study of familial and sporadic EOAD that
mutations in the APP gene only
explain a small fraction of FAD cases. The average age of disease onset in
individuals with APP gene mutations was 51.2 years. Taking into account
previous research57,
approximated that 16% of early-onset AD cases are linked to mutations in the
APP gene.
Genetic
variations in promoter sequences that modify gene expression influence
susceptibility to complex diseases. The expression levels of APP are primarily
controlled by its core promoter and the regulatory region upstream of the
5-prime end, which is linked to amyloid beta levels in AD brains. In a study
involving 427 French patients with LOAD58, identified a significant relationship between a -3102G/C SNP (rs463946)
located in the 5-prime region of the APP gene and AD. This association was
confirmed in a separate group of 502 AD cases. The C allele was protective (OR,
0.42; P = 5E-4)59. Reported on recombining the APP
gene in both normal and AD neurons, manifesting as numerous variant genomic
cDNAs. Neurons from individuals with sporadic AD exhibited a higher diversity
of genomic cDNAs, including 11 mutations associated with FAD that were not
present in healthy neurons.
APOE Associated with AD
Understanding the genetic variants of
APOE ε4 carriers has highlighted the APOE pathophysiology and resistance to AD,
offering potential therapeutic benefits. Thus, multiple genome-wide association
studies (GWAS) and meta-analyses60
demonstrated that the APOE gene (ε4 allele) remains the most common genetic
risk factor associated with sporadic AD when compared to the more prevalent ε3
allele. In addition61, found that APOE ε2 homozygosity was associated
with much lower odds of AD than APOE ε3 homozygosity. Thus, the difference
between APOE ε2 homozygosity and APOE ε4 homozygosity was even more pronounced
(0.004 [0.001- 0.014]), and APOE ε2 was linked to milder AD neuropathological
changes (i.e., fewer Aβ plaques and neurofibrillary tangles)62.
A 70-year-old Colombian woman with a
fully penetrant autosomal dominant E280A mutation in the PSEN1 gene, associated
with FAD and abundant fibrillary Aβ deposits, remained cognitively healthy
longer than expected. However, her resilience to AD was due to a rare R136S
gene mutation in APOE ε3 Christchurch63. The resistance against AD was explained as the
APOE3 R136S mutation works mechanistically by inhibiting Aβ oligomerization,
disrupting APOE binding to LDL receptors, and interfering with APOE affinity for
heparan sulfate proteoglycans. These proteoglycans are involved in the uptake
of toxic tau by neurons, which may explain the lower-than-average radioligand
uptake observed in her tau PET scan64.
MODIFIER
GENES
Recent discoveries in modifier genes
in various brain cell types have opened up new avenues for treating and halting
the advancement of numerous neurological65-67
and neurodegenerative conditions, including AD62,68.
These modifier genes can alter the expression of other target genes and
influence the penetrance, severity, or other clinically important features of
diseases caused by rare mutations in target genes. Notably, a significant
portion of FAD cases are associated with missense mutations in APP, presenilin
1 (PS1), and presenilin 2 (PS2), prompting extensive research to identify
proteins that interact with PS1 and PS2 due to their crucial roles in FAD69.
A meta-analysis study has revealed
that KLOTHO-VS heterozygosity, a polymorphism previously associated with
longevity, might reduce the increased AD risk associated with the APOE ε4
allele. A mainland Chinese cohort underwent genome sequencing, revealing nine
potential causal variants in two genes at the APOE, PVRL2, and APOC1 loci70. These
variants were found to elevate the susceptibility to AD regardless of the
presence of the APOE ε4 allele. The whole genome sequencing data stratified by
APOE genotype identified three genes significantly associated with AD in APOE4
carriers only: OR8G5 (P= 4.67E10−7), SLC24A3 (P= 2.67E−12), and IGHV3-7 (P=
9.75E-16)71.
Recently, SLC22A17 has been recognized
as a promising drug target for developing interventions to boost neurogenesis
in AD72. Conversely73, explored the genetic foundation of resilience to AD in APOE
ε4 homozygotes. They showed that CASP7 (which encodes caspase 7) rs10553596 and
SERPINA3 (which encodes α1-antichymotrypsin) rs4934-A/A polymorphisms may lower
the risk of AD73.
PROTEIN-PROTEIN
INTERACTIONS IN APOE CO-EXPRESSED GENES
We examined the network interactions
of the amyloid-beta precursor protein (APP) and co-expressed genes using the
STRING software, as shown in (Figure 2).
Interestingly, the APP network and 14 co-expressed proteins, including APOE,
APOC1, APH1A/B, PSEN1/2, PVRL2, BACE2, and NCSTN, exhibited a significantly
higher number of interactions among themselves (P< 1.0E−16) compared to what
would be expected for random proteins of similar size and distribution from the
genome. This enrichment suggests a partial biological connection between these
proteins. Notably, the previous SLC24A3, KRTCAP2, SLC22A17, and OR8G5 genes71 assigned to Alzheimer’s individuals have not
interacted or co-expressed with the APP network (Figure 2). 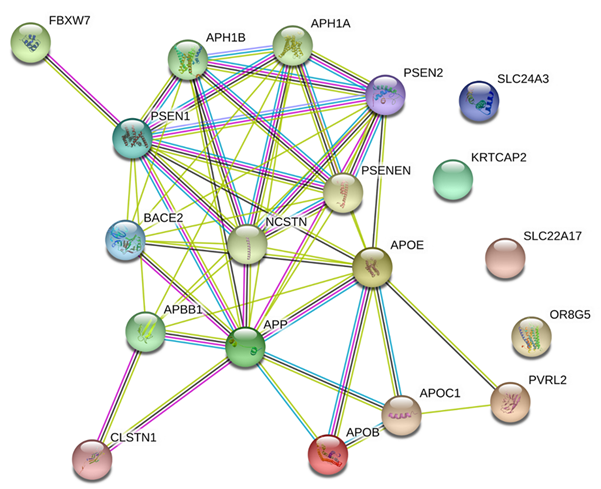

Figure 2. Protein-protein
interactions predicted by STRING (https://string-db.org/). Strong interactions
were predicted between APP and 14 co-expressed proteins. Colored nodes (n= 19)
represent proteins and the first shell of interactors (average node degree=
5.37). Edges represent specific and meaningful protein-protein associations (n=
51) (i.e., proteins jointly contribute to a shared function).
FUNCTIONAL ENRICHMENT ANALYSIS
Table 1 highlights the
biological enrichment of APP and related proteins, including APOE, APOC1,
APH1A/B, PSEN1/2, PVRL2, BACE2, and NCSTN) in biological and molecular
functions, including amyloid-beta, Notch receptor processes & tau protein
binding, and cellular components, including gamma-secretase complex (GO:0070765), triglyceride-rich, low, and chylomicron
lipoproteins. Furthermore, KEGG pathway analysis revealed the
Alzheimer’s disease, Notch signaling (hsa05010), and ‘Pfam’ revealed the
apolipoprotein C-II, A1/A4/E domains (PF05355) (Table 1).
Table 1. Functional enrichment in
APOE protein-coding gene loci network
|
GO-term |
Description |
count
in networka |
FDRb |
|
Biological
functions (GO): |
|||
|
GO:0035333 |
Notch
receptor processing, ligand-dependent |
7-Jun |
9.35E-13 |
|
GO:0033619 |
membrane
protein proteolysis |
Jul-36 |
3.83E-12 |
|
GO:0042982 |
amyloid
precursor protein metabolic process |
16-Jun |
1.01E-11 |
|
GO:0050435 |
amyloid-beta
metabolic process |
16-Jun |
1.01E-11 |
|
GO:0006509 |
membrane
protein ectodomain proteolysis |
20-Jun |
1.78E-11 |
|
GO:0042987 |
amyloid
precursor protein catabolic process |
9-May |
1.90E-10 |
|
GO:0007219 |
Notch
signaling pathway |
7/116 |
2.69E-09 |
|
GO:0016485 |
protein
processing |
7/142 |
9.43E-09 |
|
GO:0034205 |
amyloid-beta
formation |
5-Apr |
1.14E-08 |
|
GO:1905908 |
positive
regulation of amyloid fibril formation |
4-Mar |
3.28E-06 |
|
GO:0034447 |
very-low-density
lipoprotein particle clearance |
6-Mar |
7.31E-06 |
|
GO:0034382 |
chylomicron
remnant clearance |
8-Mar |
1.34E-05 |
|
GO:0043085 |
positive
regulation of catalytic activity |
10/1381 |
1.50E-05 |
|
GO:0046890 |
regulation
of lipid biosynthetic process |
5/174 |
5.25E-05 |
|
GO:1901214 |
regulation
of neuron death |
4/288 |
0.0027 |
|
GO:0007613 |
memory |
3/109 |
0.0028 |
|
GO:1904646 |
cellular
response to amyloid-beta |
26-Feb |
0.0044 |
|
Molecular
Function (GO): |
|||
|
GO:0001540 |
amyloid-beta
binding |
Apr-57 |
5.10E-05 |
|
GO:0004175 |
endopeptidase
activity |
6/399 |
0.00013 |
|
GO:0004190 |
aspartic-type
endopeptidase activity |
24-Mar |
0.00013 |
|
GO:0042277 |
peptide-binding |
5/270 |
0.00013 |
|
GO:0050750 |
low-density
lipoprotein particle receptor binding |
22-Mar |
0.00013 |
|
GO:0042500 |
aspartic
endopeptidase activity, intramembrane cleaving |
6-Feb |
0.00036 |
|
GO:0060228 |
phosphatidylcholine-sterol
O-acyltransferase activator activity |
6-Feb |
0.00036 |
|
GO:0048156 |
tau
protein binding |
18-Feb |
0.0019 |
|
Cellular
component (GO): |
|||
|
GO:0070765 |
gamma-secretase
complex |
6-May |
4.62E-11 |
|
GO:0034385 |
triglyceride-rich
plasma lipoprotein particle |
21-Apr |
5.45E-07 |
|
GO:0034363 |
intermediate-density
lipoprotein particle |
6-Mar |
3.35E-06 |
|
GO:0035253 |
ciliary
rootlet |
10-Mar |
5.69E-06 |
|
GO:0042627 |
chylomicron |
13-Mar |
8.09E-06 |
|
GO:0034361 |
very-low-density
lipoprotein particle |
20-Mar |
2.00E-05 |
|
GO:0034364 |
high-density
lipoprotein particle |
28-Mar |
4.72E-05 |
|
GO:0005794 |
Golgi
apparatus |
9/1474 |
4.75E-05 |
|
GO:0005790 |
smooth
endoplasmic reticulum |
31-Mar |
5.23E-05 |
|
GO:0043198 |
dendritic
shaft |
Mar-37 |
8.15E-05 |
|
GO:1990761 |
growth
cone lamellipodium |
3-Feb |
9.21E-05 |
|
KEGG
pathway (HSA): |
|||
|
hsa05010 |
Alzheimer's
disease |
10/168 |
3.59E-15 |
|
hsa04330 |
Notch
signaling pathway |
Jun-48 |
6.09E-11 |
|
hsa04979 |
Cholesterol
metabolism |
Mar-48 |
7.30E-05 |
|
hsa04722 |
Neurotrophin
signaling pathway |
2/116 |
0.0201 |
|
Reactome
pathway (HSA): |
|||
|
hsa-174824 |
Plasma
lipoprotein assembly, remodeling, clearance |
26-Feb |
0.00043 |
|
hsa-109582 |
Hemostasis |
3/591 |
0.0076 |
|
hsa-1430728 |
Metabolism |
4/1420 |
0.0081 |
|
Protein
domains & families (Pfam): |
|||
|
PF05355 |
Apolipoprotein
C-II |
2-Feb |
4.65E-05 |
|
PF01442 |
Apolipoprotein
A1/A4/E domain |
4-Feb |
5.81E-05 |
aProteins in the examined
network/total number of proteins.
bLog10 (observed/expected),
describing the extent of the enrichment effect.
CONCLUSION
AND FUTURE DIRECTIONS
This
review discusses the discovery, epidemiology, and gene etiology of Alzheimer’s
disease (AD). It also highlights the Alzheimer’s gender disparity common
comorbid and modifiable risk factors. Globally, AD prevalence in the general
population increases dramatically with age; it affects approximately 80% of
patients aged ≥ 75 years. Recent discoveries in modifier genes in various brain
cell types have opened up new avenues for treating and halting the advancement
of numerous neurological and neurodegenerative conditions, such as AD.
Importantly, the review’s content can guide upcoming health research on AD and
provide clinicians with evidence-based data regarding APOE and co-expressed
genes.
Funding: There is no funding to declare.
Institutional Review Board statement: NA
Authors' contributions: I thank my co-author for her equal contribution to
developing this review manuscript. The authors have read and agreed to the
published version of the manuscript.
Acknowledgments: The authors thank the Saudi Digital Library, Umm Al-Qura
University, for providing the updated scientific periodicals needed to complete
this work.
Conflicts of interest: The authors declare no conflict of interest.
REFERENCES
1. Scheltens
P, Blennow K, Breteler MM, et al. Alzheimer's disease. Lancet
2016;388(10043):505-517.
2. Scearce-Levie K, Sanchez PE, Lewcock
JW. Leveraging preclinical models for the development of Alzheimer disease therapeutics.
Nat Rev Drug Discov 2020;19(7):447-462.
3. Patterson C. World Alzheimer Report
2018. London: Alzheimer’s Disease International 2018.
4. Hebert LE, Beckett LA, Scherr PA,
Evans DA. Annual incidence of Alzheimer disease in the United States projected
to the years 2000 through 2050. Alzheimer Dis Assoc Disord 2001;15(4):169-173.
5. Long JM, Holtzman DM. Alzheimer
Disease: An Update on Pathobiology and Treatment Strategies. Cell
2019;179(2):312-339.
6. Winblad B, Amouyel P, Andrieu S, et
al. Defeating Alzheimer's disease and other dementias: a priority for European
science and society. Lancet Neurol 2016;15(5):455-532.
7. Livingston G, Huntley J, Sommerlad A,
et al. Dementia prevention, intervention, and care: 2020 report of the Lancet
Commission. Lancet 2020;396(10248):413-446.
8. Rodriguez-Vieitez E, Nielsen HM.
Associations Between APOE Variants, Tau and alpha-Synuclein. Adv Exp Med Biol
2019;1184:177-186.
9. Elhawary NA, Hewedi D, Arab A, et al.
The MTHFR 677T allele may influence the severity and biochemical risk factors
of Alzheimer's disease in an Egyptian population. Dis Markers
2013;35(5):439-446.
10. Alzheimer A. A contribution
concerning the pathological anatomy of mental disturbances in old age, 1899.
Alzheimer Dis Assoc Disord 1991;5(2):69-70.
11. McKhann
G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis
of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the
auspices of department of health and human services task force on alzheimer’s
disease. Neurology 1984;34:939-944.
12. Small GW, Rabins PV, Barry PP, et al.
Diagnosis and treatment of Alzheimer disease and related disorders. Consensus
statement of the American Association for Geriatric Psychiatry, the Alzheimer's
Association, and the American Geriatrics Society. JAMA 1997;278(16):1363-1371.
13. Chan KY, Wang W, Wu JJ, et al.
Epidemiology of Alzheimer's disease and other forms of dementia in China,
1990-2010: a systematic review and analysis. Lancet 2013;381(9882):2016-2023.
14. hara T, Ninomiya T. Trends in
dementia prevalence, incidence, and survival rate in a Japanese community.
Neurology 2017;89(18):1930-1931.
15. 2020 Alzheimer's disease facts and
figures. Alzheimers Dement 2020.
16. 2023 Alzheimer's disease facts and
figures. Alzheimers Dement 2023;19(4):1598-1695.
17. Baumgart M, Snyder HM, Carrillo MC,
Fazio S, Kim H, Johns H. Summary of the evidence on modifiable risk factors for
cognitive decline and dementia: A population-based perspective. Alzheimers
Dement 2015;11(6):718-726.
18. Chene G, Beiser A, Au R, et al.
Gender and incidence of dementia in the Framingham Heart Study from mid-adult
life. Alzheimers Dement 2015;11(3):310-320.
19. Brodaty H, Seeher K, Gibson L.
Dementia time to death: a systematic literature review on survival time and
years of life lost in people with dementia. Int Psychogeriatr 2012;24(7):1034-1045.
20. Coppus AM, Evenhuis HM, Verberne GJ, et al. Early age
at menopause is associated with increased risk of dementia and mortality in
women with Down syndrome. J Alzheimers Dis 2010;19(2):545-550.
21. Fields JA, Garovic VD, Mielke MM, et
al. Preeclampsia and cognitive impairment later in life. Am J Obstet Gynecol
2017;217(1):71-74.
22. Basit S, Wohlfahrt J, Boyd HA.
Pre-eclampsia and risk of dementia later in life: Nationwide cohort study. BMJ
2018;363:4109.
23. Andolf E, Bladh M, Moller L, Sydsjo
G. Prior placental bed disorders and later dementia: a retrospective Swedish register-based
cohort study. BJOG 2020;127(9):1090-1099.
24. Holland J, Bandelow S, Hogervorst E.
Testosterone levels and cognition in elderly men: A review. Maturitas
2011;69(4):322-337.
25. Rosario ER, Carroll J, Pike CJ.
Testosterone regulation of Alzheimer-like neuropathology in male 3xTg-AD mice
involves both estrogen and androgen pathways. Brain Res 2010;1359:281-290.
26. Jia J, Kang L, Li S, et al.
Amelioratory effects of testosterone treatment on cognitive performance
deficits induced by soluble Abeta1-42 oligomers injected into the hippocampus.
Horm Behav 2013;64(3):477-486.
27. Moffat SD, Zonderman AB, Metter EJ,
et al. Free testosterone and risk for Alzheimer disease in older men. Neurology
2004;62(2):188-193.
28. Suravarapu S, Bergstralh EJ, Farmer
SA, Knopman DS, Jacobsen SJ, Roberts RO. Dementia and low testosterone and
bioavailable testosterone levels in men: Possible increased risk. Alzheimer Dis
Assoc Disord 2006;20(3):138-140.
29. Muller M, van den Beld AW, Grobbee
DE, de Jong FH, Lamberts SW. Sex hormones and cognitive decline in elderly men.
Psychoneuroendocrinology 2009;34(1):27-31.
30. Ly M, Yu GZ, Mian A, et al.
Neuroinflammation: A Modifiable Pathway Linking Obesity, Alzheimer's disease,
and Depression. Am J Geriatr Psychiatry 2023;31(10):853-866.
31. Jayadevappa R, Chhatre S, Malkowicz
SB, Parikh RB, Guzzo T, Wein AJ. Association Between Androgen Deprivation
Therapy Use and Diagnosis of Dementia in Men With Prostate Cancer. JAMA Netw
Open 2019;2(7):196562.
32. Wingo TS, Lah JJ, Levey AI, Cutler
DJ. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch
Neurol 2012;69(1):59-64.
33. Barber IS, Braae A, Clement N, et al.
Mutation analysis of sporadic early-onset Alzheimer's disease using the NeuroX
array. Neurobiol Aging. 2017;49:215e211-e215.
34. Reitz C, Brayne C, Mayeux R.
Epidemiology of Alzheimer disease. Nat Rev Neurol 2011;7(3):137-152.
35. Lane CA, Hardy J, Schott JM. Alzheimer's
disease. Eur J Neurol 2018;25(1):59-70.
36. Pini L, Pievani M, Bocchetta M, et
al. Brain atrophy in Alzheimer's Disease and aging. Ageing Res Rev.
2016;30:25-48.
37. Chandra A, Dervenoulas G, Politis M,
Alzheimer's Disease Neuroimaging I. Magnetic resonance imaging in Alzheimer's
disease and mild cognitive impairment. J Neurol 2019;266(6):1293-1302.
38. Warren SL, Moustafa AA. Functional
magnetic resonance imaging, deep learning, and Alzheimer's disease: A systematic
review. J Neuroimaging 2023;33(1):5-18.
39. Ryan NS, Rossor MN. Correlating
familial Alzheimer's disease gene mutations with clinical phenotype. Biomark
Med 2010;4(1):99-112.
40. Bateman RJ, Aisen PS, De Strooper B, et al.
Autosomal-dominant Alzheimer's disease: a review and proposal for the
prevention of Alzheimer's disease. Alzheimers Res Ther 2011;3(1):1.
41. Ryman DC, Acosta-Baena N, Aisen PS,
et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review
and meta-analysis. Neurology 2014;83(3):253-260.
42. Lambert MA, Bickel H, Prince M, et
al. Estimating the burden of early onset dementia; Systematic review of disease
prevalence. Eur J Neurol 2014;21(4):563-569.
43. Aschenbrenner AJ, Petros J, McDade E,
et al. Relationships between big-five personality factors and Alzheimer's
disease pathology in autosomal dominant Alzheimer's disease. Alzheimers Dement
(Amst) 2020;12(1):e12038.
44. Serrano-Pozo A, Growdon JH. Is
Alzheimer's Disease Risk Modifiable? J Alzheimers Dis 2019;67(3):795-819.
45. Kuo CY, Stachiv I, Nikolai T.
Association of late life depression, (non-) modifiable risk and protective
factors with dementia and alzheimer's disease: Literature review on current
evidences, preventive interventions and possible future trends in prevention
and treatment of dementia. Int J Environ Res Public Health 2020;17(20):7475.
46. Litke R, Garcharna LC, Jiwani S,
Neugroschl J. Modifiable risk factors in alzheimer disease and related
dementias: A review. Clin Ther 2021;43(6):953-965.
47. Zhang XX, Tian Y, Wang ZT, et al. The
epidemiology of alzheimer's disease modifiable risk factors and prevention. J
Prev Alzheimers Dis 2021;8(3):313-321.
48. Passeri E, Elkhoury K, Morsink M, et
al. Alzheimer's Disease: Treatment strategies and their limitations. Int J Mol
Sci 2022;23(22):13954.
49. Theuns J, Del-Favero J, Dermaut B, et
al. Genetic variability in the regulatory region of presenilin 1 associated
with risk for Alzheimer's disease and variable expression. Hum Mol Genet
2000;9(3):325-331.
50. Wragg M, Hutton M, Talbot C. Genetic
association between intronic polymorphism in presenilin-1 gene and late-onset
Alzheimer's disease. Alzheimer's Disease Collaborative Group. Lancet
1996;347(9000):509-512.
51. Guerreiro RJ, Lohmann E, Kinsella E
et al. Exome sequencing reveals an unexpected genetic cause of disease: NOTCH3
mutation in a Turkish family with Alzheimer’s disease, Neurobiology of Aging
2012;33(5):17-23.
52. Khanahmadi M, Farhud DD, Malmir M.
Genetic of alzheimer's disease: A narrative review article. Iran J Public
Health 2015;44(7):892-901.
53. Achouri-Rassas A, Ali NB, Fray S, et
al. Novel presenilin 1 mutation (p.I83T) in Tunisian family with early-onset
Alzheimer's disease. Neurobiol Aging 2015;36(10):2904.
54. Roher AE, Maarouf CL, Kokjohn TA.
Familial Presenilin Mutations and Sporadic Alzheimer's Disease Pathology: Is
the Assumption of Biochemical Equivalence Justified? J Alzheimers Dis
2016;50(3):645-658.
55. Glenner GG, Wong CW. Alzheimer's
disease: Initial report of the purification and characterization of a novel
cerebrovascular amyloid protein. Biochem Biophys Res Commun
1984;120(3):885-890.
56. Tanzi RE, Vaula G, Romano DM, et al.
Assessment of amyloid beta-protein precursor gene mutations in a large set of
familial and sporadic Alzheimer disease cases. Am J Hum Genet
1992;51(2):273-282.
57. Raux G, Guyant-Marechal L, Martin C,
et al. Molecular diagnosis of autosomal dominant early onset Alzheimer's disease:
an update. J Med Genet 2005;42(10):793-795.
58. Guyant-Marechal L, Rovelet-Lecrux A,
Goumidi L, et al. Variations in the APP gene promoter region and risk of
Alzheimer disease. Neurology 2007;68(9):684-687.
59. Lee MH, Siddoway B, Kaeser GE, et al.
Somatic APP gene recombination in Alzheimer's disease and normal neurons.
Nature 2018;563(7733):639-645.
60. Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic
meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and
implicates Abeta, tau, immunity and lipid processing. Nat Genet
2019;51(3):414-430.
61. Reiman EM, Arboleda-Velasquez JF, Quiroz YT, et al.
Exceptionally low likelihood of Alzheimer's dementia in APOE2 homozygotes from
a 5,000-person neuropathological study. Nat Commun 2020;11(1):667.
62. Serrano-Pozo A, Das S, Hyman BT. APOE and Alzheimer's
disease: advances in genetics, pathophysiology, and therapeutic approaches.
Lancet Neurol 2021;20(1):68-80.
63. Arboleda-Velasquez JF, Lopera F,
O'Hare M, et al. Resistance to autosomal dominant Alzheimer's disease in an
APOE3 Christchurch homozygote: a case report. Nat Med 2019;25(11):1680-1683.
64. Belloy ME, Napolioni V, Han SS, et
al. Association of Klotho-VS Heterozygosity With Risk of Alzheimer Disease in
Individuals Who Carry APOE4. JAMA Neurol 2020;77(7):849-862.
65. Elhawary NA, AlJahdali IA, Abumansour IS, et al. Genetic
etiology and clinical challenges of phenylketonuria. Hum Genomics
2022;16(1):22.
66. Mangano GD, Riva A, Fontana A, et al. De novo GRIN2A variants
associated with epilepsy and autism and literature review. Epilepsy Behav
2022;129:108604.
67. Elhawary NA, AlJahdali IA, Abumansour
IS, et al. Phenotypic variability to medication management: an update on fragile
X syndrome. Hum Genomics 2023;17(1):60.
68. Jain N, Chen-Plotkin AS. Genetic
Modifiers in neurodegeneration. Curr Genet Med Rep 2018;6(1):11-19.
69. Yang Y, Bagyinszky E, An SSA. Presenilin-1 (PSEN1) Mutations:
Clinical Phenotypes beyond Alzheimer's Disease. Int J Mol Sci 2023;24(9):8417.
70. Zhou X, Chen Y, Mok KY, et al.
Non-coding variability at the APOE locus contributes to the Alzheimer's risk.
Nat Commun 2019;10(1):3310.
71. Ma Y, Jun GR, Zhang X, et al. Analysis of whole-exome
sequencing data for alzheimer disease stratified by APOE genotype. JAMA Neurol
2019;76(9):1099-1108.
72. Siddiqui T, Cosacak MI, Popova S, et
al. Nerve growth factor receptor (Ngfr) induces neurogenic plasticity by
suppressing reactive astroglial Lcn2/Slc22a17 signaling in Alzheimer's disease.
NPJ Regen Med 2023;8(1):33.
73. Huq AJ, Fransquet P, Laws SM, et al.
Genetic resilience to Alzheimer's disease in APOE epsilon4 homozygotes: A systematic
review. Alzheimers Dement 2019;15(12):1612-1623.