Combination of Hurler Syndrome and Nasosinus Polyposis: Case Report and Review of the Literature
Abstract
Mucopolysaccharidosis (MPS) type 1 is characterized by
a heterogeneous clinical spectrum, a progressive evolution and multisystemic
manifestations including ENT. Early recognition of MPS by otolaryngologists
play an increasingly important role in the multidisciplinary approach to
diagnosis and management of many children with MPS. In addition to symptomatic
measures, current treatments for MPS I include enzyme replacement therapy and
hematopoietic stem cell transplantation alone or in combination.
In this context, we report the case of a child
suffering from Haller's syndrome who underwent surgery in our department for
nasosinus polyposis and adenoid vegetations, thus improving respiratory comfort
and guiding the diagnosis of Hurler syndrome.
Keywords: Hurler
Syndrome; ENT symptoms; Case Report; Surgery
Introduction
Head and neck disorders affect the majority of
mucopolysaccharidosis patients. Symptoms such as sleep apnea, frequent
respiratory and ear infections, chronic nasal discharge and enlarged tonsils
and adenoids, may be indicative of MPS disease. Therefore, a more complete
diagnostic search will enable us to establish an early diagnosis and,
consequently, provide adequate care, enabling these patients to enjoy a better
quality of life and longer life expectancy1,2.
In this perspective, we report the case of a
3-year-old child who consulted our department for chronic nasal obstruction,
bulging and sleep apnea. The patient underwent a CT scan of the nasal cavity
showing nasosinus polyposis and hypertrophy of the adenoid vegetations, before
deciding to undergo surgery.
Case
report
We report the case of a 3-year-old child with no
particular medical history, admitted to our ENT department for bilateral nasal
obstruction.
The history of the disease dates back 2 years, with
the onset of bilateral nasal obstruction associated with rhinorrhea, bulging
and sleep apnea. The patient's general condition was good.
On ENT physical examination, anterior rhinoscopy
revealed complete filling of both nasal cavities by polyps extending to the
floor of the nasal cavities. Endobuccal examination revealed bilateral
tonsillar hypertrophy.
A CT scan of the nasal cavities showed a hypodense,
homogeneous, confluent process filling the maxillary sinuses, frontal sinuses,
ethmoidal cells, sphenoidal sinus and nasal cavities, in favor of bilateral
nasosinus polyposis. There is also a significant hypertrophy of the posterior
soft tissues of the cavum, leading to obstruction of the upper airway (Figures1 and 2).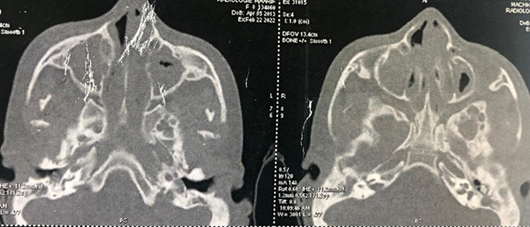
Figure 1: Axial- section CT scan of
the nasal cavities showing total filling of the maxillary, ethmoidal, frontal
and sphenoidal sinuses and nasal cavities.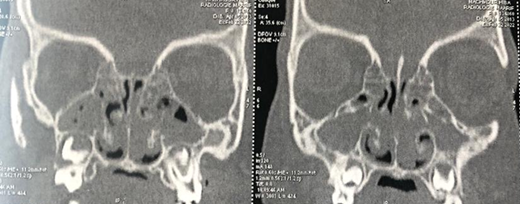
Figure 2: CT scan of the nasal
cavities in coronal section, showing complete filling of the maxillary,
ethmoidal, frontal and sphenoidal sinuses and nasal cavities.
On this basis, the therapeutic decision taken at the
staff meeting was to perform a bilateral polypectomy, a minimal inferior
turbinectomy (just the proximal part of the inferior turbinate), a bilateral
middle meatotomy, a tonsillectomy and an endobuccal removal of adenoid
vegetation.
The patient was then referred to the pediatrics
department for further follow-up. On specialized pediatric examination, based
on our ENT observations, the diagnosis of Hurler syndrome was made. In terms of
ENT, during post-operative consultations, respiratory progress was excellent,
with improved respiratory comfort without swelling or sleep apnea.
Discussion
A group of hereditary metabolic diseases known as
mucopolysaccharidosis is caused by a deficiency in the specific enzymes
involved in the lysosomal degradation of glycosaminoglycans (GAGs). The type I
mucopolysaccharidoses (MPS 1) are due to a lack of alpha-L-iduronidase, the
enzyme responsible for the hydrolysis of heparan sulfate and dermatan sulfate.
A German pediatrician, Hurler, reported the first cases of MPS I in 19191,2.
Type 1 mucopolysaccharidosis can be classified into
three different forms depending on the mutations, Hurler's disease and Scheie's
disease, representing the two extremes of the spectrum of severity, and
Hurler-Scheie disease which is the intermediate phenotype1.
Hurler syndrome is the most severe form of MPS,
leading to death in childhood. The child appears normal at birth. Diagnosis is
usually made between 4 and 18 months of age, based on the association of
macroglossia, skeletal deformities, hepatomegaly accompanied by umbilical and
inguinal hernias, and recurrent infections of the ears and upper and lower
airways3.
If there is a strong clinical suspicion, a
quantitative and qualitative study of urinary GAGs is often carried out as a
first line of investigation, and if positive, can help to orientate the
diagnosis of MPS I. This is confirmed by measuring alpha-L-iduronidase enzyme
activity in leukocytes and fibroblasts4.
A multidisciplinary team is required to manage the
disease, including orthopedic surgeons, neurosurgeons, cardiologists,
otorhinolaryngologists, physiotherapists and others. Early diagnosis of MPS is
essential to enable patients to benefit from rapid therapeutic intervention5.
A genetically-engineered analogue of human alpha-
L-iduronidase, laronidase, is the enzyme replacement therapy used in the
treatment of MPS I, with a dosage of 100 IU/kg6.
Respiratory infections, facial dysmorphia and hernia
were the most frequent reasons for consultation, according to7. All MPS I patients, and particularly those
with the Hurler phenotype, are at risk of developing severe respiratory failure
as a result of pulmonary restrictive disease, sleep apnea and/or asthma8.
ENT doctors are frequently in contact with patients
referred for ENT symptoms before the diagnosis of MPS is made, which enables
them to make an early diagnosis of this disease. Symptoms such as sleep apnea,
frequent ENT and respiratory infections, macroglossia, hypertrophy of the
adenoids and tonsils often, irregular nasal septum and turbinate hypertrophy
appear several years before the definitive diagnosis of MPS. Obstructive
symptoms are at first more pronounced in the upper airways, with tracheobronchial
manifestations occurring later. Because of the small number of patients,
however, no conclusions can be drawn as to the prevalence and severity of
respiratory problems for MPS9,10.
The resulting hypertrophy and accumulation of GAGs in
the adenoids and tonsils have made these structures common targets for surgical
intervention. Adenoidectomy and tonsillectomy provide only temporary relief of
upper airway obstruction, also risks are generally higher in a child with MPS,
including postoperative hemorrhage, airway edema and extubating failure. Both
chronic rhinosinusitis and chronic otitis media may develop11,12.
It should also be noted that, in addition to the poor
prognosis associated with delayed diagnosis, the fact that MPS patients undergo
surgical procedures before being diagnosed is a major worry, given that the
anesthetic risk is extremely high in these patients, due to deformities of the
larynx, trachea and lower respiratory tract13.
In terms of outcome, Lin et al. found that enzyme
replacement therapy in MPS helped to reduce cardiac hypertrophy, particularly
when administered at an early age, but has little effect on valvular damage.
Without replacement therapy, Bousssof's results show that the general condition
of most patients deteriorates, with progressive physical deterioration and loss
of quality of life14,15.
Conclusion
Mucopolysaccharidosis type 1 is responsible for
multisystem damage that progressively worsens gradually with age. This calls
for multidisciplinary management based on specific treatment and symptomatic
measures. A wide range of ENT symptoms appear in the early stages of MPS,
including rhinosinusitis, macroglossia, adeno-tonsillar hypertrophy, nasal
obstruction, OSA, progressive respiratory disorders and hearing loss.
Otorhinolaryngologists should be aware of MPS, particularly in young children
(2-3 years) with an indication for adenotonsillectomy.
References
1. Chalès
G. Guggenbuhl P. Mucopolysaccharidoses et oligosaccharidoses. EMC-Rhumatologie
Orthopédie 2004;1(5):395-405.
2. Muenzer J. The mucopolysaccharidosis: a heterogeneous
group of disorders with variable pediatric presentations. J
Pediatr 2004;144:27-34.
3. d’Orphanet
LC, Rares SM. Prévalence des maladies rares: Données bibliographiques. Janvier, Numéro 2019;2.
4. Muenzer J, Wraith JE, Clarke LA, the International
Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I.
Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics
2009;123(1):19-29.
5. Sawamoto K, Stapleton M, Alméciga ‑Díaz C, et al.
Therapeutic options for mucopolysaccharidoses: Current and emerging treatments.
Drugs 2019;79(10):1103-1134.
6. Haute Autorité de Santé.
Mucopolysaccharidose de type I: Protocole national de diagnostic et de soins
Guide -affection de longue Ce document est disponible sur 2007.
7. Kuiper GA, Meijer OLM, Langereis EJ, Wijburg FA.
Failure to shorten the diagnostic delay in two ultra-orphan diseases
(mucopolysaccharidosis types I and III): potential causes and implications.
Orphanet J Rare Dis 2018;13(1):2.
8. Faverio P, Stainer A, De Giacomi F, et al. Molecular pathways
and respiratory involvement in lysosomal storage diseases. Int J Mol Sci 2019;20(2):327.
9. Bianchi PM, Gaini R, Vitale S. ENT and
mucopolysaccharidoses Ital J Pediatr 2018;44:127.
10. Berger KI, Fagondes FC, Giugliani R, et al.
Respiratory and sleep disorders in mucopolysaccharidosis. J Inherit Metab Dis
2013;36:201-210.
11. Muhlebach MS, Wooten W, Muenzer J. Respiratory
manifestations in mucopolysaccharidoses. Paediatr Resp Rev 2011;12(2):133-138.
12. Mesolella
M, Cimmino M, Cantone E, et al. Management
of otolaryngological manifestations in mucopolysaccharidoses: our experience.
Acta Otorhinolaryngol Ital 2013;33:267-272.
13. Torres DdeA, Barth AL, Valente MPdeM, Mello PP,
Horovitz DDG. Otolaryngologists and the early diagnosis of
mucopolysaccharidoses: A cross-sectional study. Diagnostics 2019;9(4):187.
14. Lin HY, Chuang CK, Chena MR, et al. Cardiac structure
and function and effects of enzyme replacement therapy in patients with
mucopolysaccharidoses I, II, IVA and VI. Mol Genet Metab 2016;117(4):431-437.
15. Elmehdi B. Les
mucopolysaccharidoses de type I : A propos de 10 cas. Faculté de médecine et
pharmacie de Rabat 2012.