Congenital Pulmonary Airway Malformation (CPAM). A Case Report
Abstract
Congenital bronchopulmonary lesions are rare pathologies derived from airway developmental defects. This group of heterogeneous conditions demonstrates common characteristics on imaging, potentially making diagnosis difficult. Most of these lesions are detected pre-natally and specialists must be familiar with their aspect in different imaging methods.
This study presents a pregnancy of 17 weeks and a fetal diagnosis of cystic right pulmonary lesion found on routine ultrasound. During the pregnancy, follow-up ultrasounds and a magnetic resonance (mr) were performed to assess the lesion's growth and characteristics. The imaging studies showed progressive growth of the lesion, causing mediastinal displacement.
At birth, the newborn exhibited respiratory symptoms. Imaging studies revealed a large cystic lesion in the right upper lobe, which was later confirmed to be a type i cpam. The patient underwent surgery and the postoperative recovery was favorable.
This case underscores the importance of prenatal diagnosis and careful monitoring of cpams, as well as the need for timely surgical intervention in symptomatic cases. Sequential evaluation through ultrasound and mri is crucial for determining the appropriate clinical and surgical management.
Keywords: congenital bronchopulmonary malformation; cpam ratio; congenital airway malformation; fetal development; prenatal diagnosis.
Introduction
Congenital bronchopulmonary
malformations represent a broad range of pathologies with characteristic
features for each of them. This group includes pulmonary sequestration,
congenital pulmonary airway malformation (cpam), bronchogenic cyst, congenital
pulmonary overinflation and pulmonary atresia. It is considered that most
pathologies on this group exhibit origin from an early altered development of
pulmonary airway with subsequent anomalies of the lung structure1. Diagnosis for most of the
lesions on this group is made with ultrasound, performed antenatally or in
early peri-natal interval. These lesions tend to reduce its size during third
trimester and become difficult to detect at birth. This is why some of these conditions
could not be detected post natally2,3. Newborn images can be
normal or display subtle findings, specially on conventional radiology studies.
Furthermore, some indexes like the cvr (cpam volume ratio) are developed to be
applied on antenatal ultrasound with an important prognostic value. The
analysis of the index values along the gestational period defines the
management of the patient, with options like surveillance and surgical
intervention pre or post natally3,4.
Case
presentation
A
44year old women with previous medical history of a spontaneous abortion.
Current pregnancy was achieved by egg donation. At 17 weeks of gestational age
the ultrasound study revealed an unilocular cystic lesion on the right lung
with size of 15.8 x 12.4 x 15.5 mm and a volume of 1.4cc. The lesion caused
mediastinal shift to contralateral. Doppler color did not show internal
vascularity or afferent vascularity (figure 1). The cpam volume ratio
(cvr), used commonly for the evaluation of antenatal pulmonary lesions,
reflected a value of 0.12.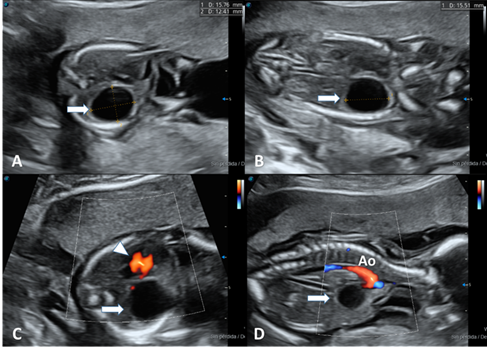
Figure1: obstetric ultrasound showing a unilocular
cystic image (arrow) with echogenic borders in the topography of the right
lung. A and c transverse views. B and d longitudinal view. On color doppler
evaluation, no vascularization is present. Arrow tip: heart, ao: aorta
The
amniotic fluid volume was normal and no signs of fetal hydrops were found in
pre-natal studies. Ultrasound control studies along the pregnancy showed these
results: 18 weeks (cvr: 0.21), 21 weeks (cvr: 0.45), 28 weeks (cvr: 0.57) and
35 weeks (cvr: 0.42) (figures 2,3).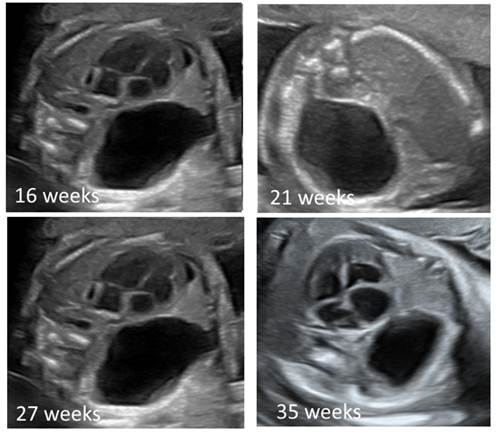
Figure 2: obstetric
ultrasounds in four-chamber view showing the evolution of the unilocular cystic
image with echogenic borders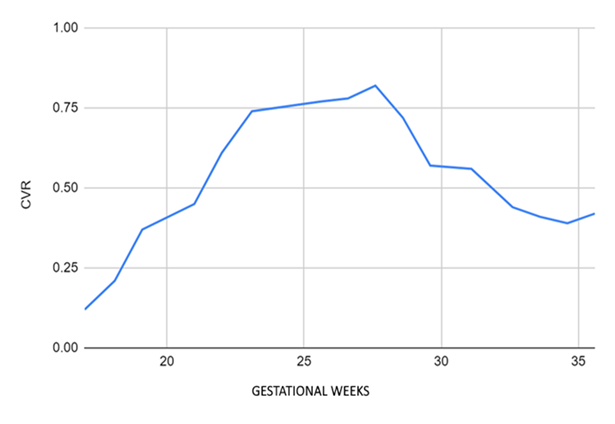
Figure 3: evolution of the cyst volume ratio. Cvr (cpam volume ratio). Volume lesion/head circumference ratio.
Differential diagnosis included bronchogenic cyst, congenital pulmonary airway malformation and pulmonary sequestration. Mr without paramagnetic contrast was performed at 28 weeks. It demonstrated increased size of the lesion (33.8 x 48.5 x 26.6mm) with a volume of 21cc. The calculated remnant pulmonary volume on both thoracic cavities was 22.3cc. This value corresponded to 41.3% of the expected percentage for this gestational age (figure 4). Mediastinal shift persisted and no signs of fetal hydrops were found. The mr study did not show other related malformations.
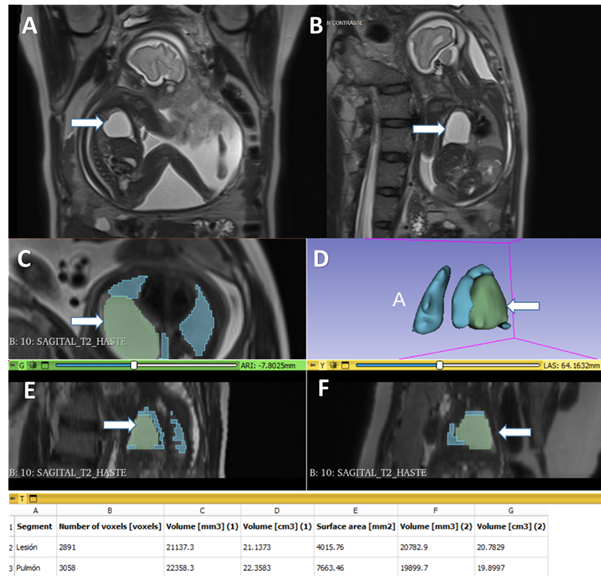
Figure 4: magnetic resonance imaging without contrast at 28 weeks, showing t2-weighted images in a (sagittal) b(coronal) view of the fetus. It is possible to recognize a hyperintense round lesion in the topography of the right lung (arrows). D, e and f. Total fetal lung volumetry, residual and of the cystic lesion.
Delivery was performed via cesarean section at 37.5 weeks of gestation. Newborn was male, weight 3235g, apgar 7-8. He received supplementary oxygen with a fio2 of 25-30% until three days post-delivery. Initial chest x-ray showed an ill-defined opacity on the upper segment of the right lung. After this time the newborn clinical evolution was favorable with expectant management. There was not need for supplemental oxygen and there were no signs of respiratory insufficiency. Echocardiogram did not reveal structural abnormalities.
After the first month of life the patient was
admitted in the emergency room with signs of respiratory insufficiency
(tachypnea, subcostal and intercostal retraction, generalized cyanosis),
irritability and difficulty feeding. Chest x-ray revealed an important
radiolucent lesion on the right hemi thorax. The lesion was wall thinned, well
defined and caused important contralateral mediastinal shift. Chest ct with
angiogram exhibited a voluminous cystic mass in the upper lobe of the right
lung, size 81 x 67 x 58mm. The compressive effect of the lesion caused partial
atelectasis of the lower lobe with diffuse increase in its density. There were
signs of air entrapment in the middle lobe and compression of the intermediary
bronchus. There was not systemic vascularity recognized on the ct-angiogram (figure 5).
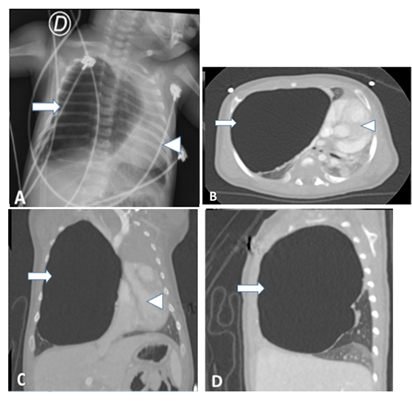
Figure 5: a. Chest x-ray one month old after birth. Image
shows an air-filled cystic image (arrow) that displaces the mediastinum (arrow
tip) to the left. B, c and d ct scan images with lung window in axial, coronal
and sagittal projections demonstrating an unilocular cystic image with air
content compressing the adjacent lung parenchyma and produces mediastinal
deviation. Intravenous contrast did not show internal or systemic vascularity.
Emergency surgery demonstrated a voluminous cystic mass occupying the upper right pulmonary lobe. There was also signs of compromised vascularity of the lateral segment of the middle lobe secondary to mechanical compression caused by the cystic mass. A total upper lobectomy and an atypical segmentectomy were performed.
Histopathologic study exhibited a cyst with
fibromuscular wall and luminal epithelium of ciliated cylindrical cells. It was
surrounded by normal pulmonary parenchyma and smaller cysts with similar
characteristics to the main one. These findings were compatible with the
diagnosis of congenital pulmonary airway malformation type i (figure 6). Post-operative evolution of the patient was
favorable. The remaining pulmonary parenchyma of the right lung expanded
without complications. Radiological follow-up demonstrated complete resolution
of the lesion.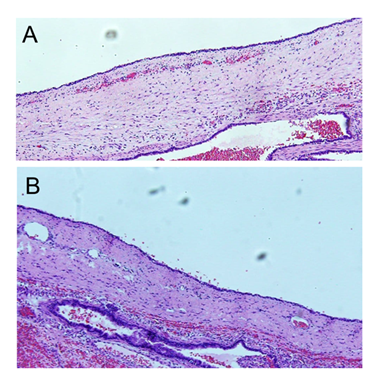
Discussion
Congenital pulmonary airway malformation (cpam),
known previously as cystic adenomatoid malformation, is a rare congenital
abnormality. Nevertheless, it is the most frequent of the congenital
bronchopulmonary malformations. It has a frequency of 25%, with an incidence of
1:25.000 – 1:35.000. First described in 1949 by ch'in y tang and later
classified by stoker4,5.
- type
0: previously known as acinar dysplasia. It is a lesion located in the main
tracheobronchial structures. It is typically solid, with a scarce cystic
component that measures less than 0.5 cm in size. This lesion is the least
common and has a high mortality rate during the neonatal period6,7.
- type
1: is the most common type with a percentage of 60-70% of all cpam. The main
feature of this lesion include a cystic lesion, sole or multiple, measuring
more than 2cm and less than 10cm. The size is considered a potential element to
differentiate this lesion from type 4 cpam. Histologically its wall is lined by
columnar epithelium. The prognosis of this type of lesion is good1,6-8.
- type
2: it originates from terminal bronchi. This type of lesion represents 15-30%
of al cpam. It is a mixed cystic-solid lesion with a cystic component less than
2cm. It associates frequently with other fetal malformations like
tracheoesophageal fistula, renal agenesis, bowel atresia and diaphragmatic
hernia6,9,10.
- type
3: it derives from alveolar tissue. It represents 8% of cases. This type of
lesion is mainly solid wit cystic component less than 5mm or only recognizable
at microscopic level. The main differential diagnosis is pulmonary
sequestration6-10.
- type 4: it arises from the most distal pulmonary acini. This lesion presents as cysts of size greater than 10cm. The wall can be thick, associated with internal septa and occasionally spontaneous pneumothorax. Histologically its wall is made of type i and ii pneumocytes. It is also associated with the development of pulmonary blastoma. This lesion is identified at birth or during early childhood. From the point of view of imaging it can be indistinguishable of cpam type 16-10.
Diagnosis of cpam is usually done with ultrasound during the second trimester because it is difficult to recognize before 17 weeks of gestation. This consideration is applicable specially for cpam type 1 and 3, which are commonly detected on pre-natal period. A more detailed evaluation requires methods like mr to better define location, size, local compromise, mass effect and internal architecture of the lesion1,3. Cpam potentially grow along the pregnancy, reaching their maximum size between 20 and 26 weeks. After this time some of this lesions tend to reduce their size or even disappear1,3,8. Size of the lesion is a determinant factor for the evolution and prognosis of these patients. An important parameter to evaluate the risk of negative outcomes is the cpam volume ratio (cvr). This index compares volume of the lesion with the value of cephalic perimeter. It allows to predict the risk for fetal hydrops, specially if its vale is more than 1.6. Identifying fetal hydrops requires urgent management or labor induction11,12.
When pulmonary malformations are important in size or produce clinical symptoms, it is necessary to complete with imaging evaluations post-natally. Main radiological evaluations include x-rays, ultrasound and computed tomography3. Chest x-ray is the first method used for initial evaluation13. Patients with an antenatal diagnosis of pulmonary malformation, initial chest x-ray could appear normal given that lesions tend to reduce size during third trimester and in early peri-natal period. Visibility of these lesions can be reduced because of persistence of fetal liquid inside them. Initial radiographic appearance depends on the size and content of the lesion. They can be radiopaque because of persistence of fetal fluid distal to abnormal obstructed airways. Or, they can demonstrate a cystic appearance with air density, coming from patent airways8. Post-natal ultrasound can visualize the lesion specially if it does not contain air inside it8,11.
Postnatal mr can potentially be used for better differentiation between solid and cystic lesions compared to ct. However, it has its own limitations: requirement of anesthesia and lung atelectasis derived from this procedure, prolonged study times and the inherent difficult evaluation of the normal pulmonary parenchyma through this method1,3. Alternatively, ct is a fast-imaging method and commonly does not require anesthesia. Angiogram ct offers important information about vascular supply of the lesion and the compromise of adjacent structures. However, it uses ionizing radiation that is increasingly harmful especially for children. Use of mr or ct relies on the criteria of the practitioner and the consultant radiologist, the availability of the method and the consent of the family1-3. Some authors consider that asymptomatic patients in the newborn period with perfectly defined prenatal categorized lesions can be evaluated by imaging after 3-6 months of life. After this time and with all the information obtained it is possible to consider expectant or surgical management. The main objective of imaging evaluation is to determine small lesions with focal bronchial atresia, mucous impaction o focal overinflation, suitable for expectant management. Conversely voluminous lesions, multi-cystic lesions and those with arterial vascular supply are amenable to surgical management1,2. Other features affecting the management of the lesion are extension and location. About this, it is considered that upper lobe lesions tend to be more symptomatic in comparison with other locations1.
Differential diagnosis between cpam and other bronchopulmonary malformations can be difficult. Bronchopulmonary sequestration is a non-functional pulmonary mass with systemic vascular supply and commonly without patent connection with the tracheobronchial tree. Imaging aspects include a solid heterogeneous mass with subtle cystic component. It usually locates in lower lobes1,2,8. This type of lesion is divided as intralobar and extralobar. One prominent difference between these is the venous drainage to the pulmonary veins or the azygos vein respectively1,2,8. Other differential aspect between cpam and pulmonary sequestration is that the former can have arterial supply form the pulmonary arteries. In contrast, pulmonary sequestration receives vascular supply from the thoracic aorta1,14.
Bronchogenic cyst usually contains mucin-liquid and locates near the mediastinum. It is possible to find it in the pulmonary parenchyma but this is rare. Mucinous contents produce altered signal intensity on mr and can also contain calcium foci1-3. As with pulmonary sequestration, it commonly does have connection with tracheobronchial tree.
Congenital pulmonary overinflation derives form and abnormal developed bronchus or extrinsic airway compression. This pathology tends to produce a cranio-caudal enlargement and expansion of the affected lung parenchyma. This causes more atelectasis of the normal adjacent lung parenchyma and less mediastinal shift8-15. Meanwhile cpam is more likely to grow toward the pulmonary hilum and produce mediastinal shift. Cpam also tend to displace peripherally vascular markings, while congenital pulmonary overinflation causes lesser displacement of vascular structures.
Bronchial atresia is commonly considered an associated finding in multiple bronchopulmonary congenital pathologies, especially in congenital pulmonary overinflation and cpam. Bronchial atresia imaging features include a focal zone of reduced diameter associated with mucous plugs and focal zones of pulmonary parenchyma hyperinflation1,3.
Other pathologies to be taken into consideration for the differential diagnosis of bronchopulmonary malformations include pulmonary neoplasms2,3. Congenital pulmonary neoplasms are usually found on the third trimester of gestation, conversely to congenital pulmonary malformations which are mainly identified on the second trimester1. Congenital lung tumors include fibrosarcoma, myofibroblastic tumor and mesenchymatous hamartoma. These lesions present a heterogeneous aspect due to mucinous or mesenchymatous component with multiple and inmmature cells1,2,8. Cystic pulmonary blastoma is an infrequent lung tumor. It is typically diagnosed in the third trimester, located peripherally on the lung, with multiple cystic foci, thickened walls, associated pneumothorax and familiar history of tumors3.
Histologically cpam type 1 manifest as cystic structures, unique or multiple, surrounded by normal pulmonary parenchyma. Cysts are lined by columnar or cuboid epithelium and stroma of fibrous or fibromuscular tissue. Cysts originate from abnormally enlarged bronchus associated with irregular growing of epithelial cells. Unlikely other bronchopulmonary malformations, cpam type 1 does not demonstrate normal internal alveolar architecture1,2. Cpam type 4 demonstrates cystic component lined by type 1 and 2 pneumocytes. Usually this lesion presents less fibrous tissue compared to cpam type 1.
In the present case the pulmonary lesion was recognized during the second trimester of pregnancy. The finding was an unilocular cyst, well defined, without septa, solid component or internal vascularity. Control prenatal ultrasounds revealed reduction on its size with a cvr value of 0.57 at 28 weeks and 0.42 at 35 weeks. Its is considered that a cvr value greater than 0.39 between 25 and 29 weeks, additionally with mediastinal shift, are predictive factors for surgical management in the two years of life12. This outcome was found in the case presented. Prenatal mr confirmed location of the lesion in the upper right lobe with important contralateral mediastinal shift. None of the imaging control studies revealed fetal hydrops and a conservative approach was decided
Taking into consideration the data pre and post nataly, the main diagnosis proposed was cpam type 1. Another diagnostic option like bronchogenic cyst was less likely because this kind of lesion locates in the mediastinum in 70% of cases and causes little mediastinal shift2. There was not systemic vascularity of the lesion, confirmed finding on the post-natal ct angiogram, which discards diagnosis of pulmonary sequestration. Bronchial atresia and congenital lung overinflation appear on antenatal ultrasound as changes in pulmonary parenchyma echogenicity and not as cystic lesions. Although cpam type 4 can be indistinguishable from cpam type 1 through imaging methods, there are some features that can help to make a differential diagnosis. In the presented case the cystic lesion was recognized in second trimester, was located more centrally in the lung, reduced it size in the third trimester, did not show internal septa and there was not pneumothorax associated7. Pleuropulmonary blastoma usually is diagnosed in later periods of gestational period or in early pregnancy. It also associates with multilocular cystic nephroma16, which was not found in the presented case. Histologically the lesion presented a columnar epithelial lining and fibromuscular wall. All these features confirm the diagnosis of cpam type 1, which was also the most accepted diagnosis all along the patient follow up.
Management of asymptomatic patients with cpam remains controversial. However, many guides recommend surgical resection considering risk of infection and, less probably, future development of pulmonary tumors. In the presented case emergency surgical management was necessary for the appearance of respiratory distress symptoms in the first month of life. Surgery usually is performed between the second and sixth month of life to reduce anesthetic risk in neonatal period17. Final prognosis depends on the size of the lesion, associated pulmonary hypoplasia or the presence of other congenital abnormalities18.
Conclusion
Bronchopulmonary malformations are a
heterogeneous group of pathologies derived from abnormal development of airways
during fetal life. Cpam is the most frequent congenital lung malformation.
Nevertheless, it can be difficult to distinguish form other lung pathologies.
The specific type of congenital lung malformation can be properly characterized with data of antenatal and post-natal imaging studies. When lesions have not been studied intra-utero, post-natal characterization can be really difficult or even delay the diagnosis until adult life, especially when they are asymptomatic. Cpam volume ratio (cvr) has demonstrated being an important prognostic factor in the future clinical outcome of the patients. This is confirmed by the fact the studied patient required surgical management in the first month of life. This information is potentially useful for making changes in the management of this group of patients.
References
3. Newman b. Magnetic resonance imaging for congenital
lung malformations. Pediatr radiol 2022;52(2):312-322.
4. Ch'in ky, tang my. Congenital adenomatoid
malformation of one lobe of a lung with general anasarca. Arch pathol (chic)
1949;48(3):221-229.
5. Stocker jt. The respiratory
tract, in: stocker jtdehner lp, eds, pediatric pathology, 2nd ed, philadelphia,
pa: lippincott, williams and wilkins 2001;445-517.
6. Phillips p. Congenital
pulmonary airway malformation: a case study and case comparison. J diagnostic
medical sonography 2016;32(5):294-298.
7. Stocker jt, madewell je, drake rm. Congenital
pulmonary airway malformation: a new name and an expanded classification of
congenital cystic adenomatoid malformation of the lung. Histopathology
2002;41(2):424-431.
8. Kao sw, zuppan cw, young lw. Airp best cases in
radiologic-pathologic correlation: type 2 congenital cystic adenomatoid
malformation (type 2 congenital pulmonary airway malformation). Radiographics.
2011;31(3):743-748.
9. Griffin n, devaraj a, goldstraw p, bush a, nicholson
ag, padley s. Ct and histopathological correlation of congenital cystic
pulmonary lesions: a common pathogenesis? Clin radiol 2008;63(9):995-1005.
10. Stocker jt. Cystic lung
disease in infants and children. Fetal pediatr pathol 2009;28(4):155-184.
11. Jordan ka. Sonographic
appearance of congenital pulmonary airway malformations. J diagnostic medical
sonography 2016;32(4):237-239.
12. Peters ncj, hijkoop a,
hermelijn sm, et al. Prediction of postnatal outcome in fetuses with congenital
lung malformation: 2-year follow-up study. Ultrasound obstet gynecol
2021;58(3):428-438.
13. Newman b. Congenital
bronchopulmonary foregut malformations: concepts and controversies. Pediatr
radiol 2006;36(8):773-791.
14. Langston c. New concepts in the pathology of
congenital lung malformations. Semin pediatr surg
2003;12(1):17-37.
15. Álvarez
barrial m, nava hurtado de saracho fb, bueno jiménez a, et al. Congenital pulmonary airway
malformation (cpam) mimicking an spontaneous pneumothorax in a newborn. Malformación
pulmonar congénita simulando un neumotórax espontáneo en un recién
nacido. Cir
pediatr 2021;34(4):207-210.
16. Azizkhan rg, crombleholme tm.
Congenital cystic lung disease: contemporary antenatal and postnatal
management. Pediatr surg int 2008;24(6):643-657.
17. Chuang s, sugo e, jaffe a. A
review of postnatal management of congenital pulmonary airway malformations.
Fetal matern med rev 2009;20(3):179-204.
18. Priest jr, williams gm, hill
da, dehner lp, jaffé a. Pulmonary cysts in early childhood and the risk of
malignancy. Pediatr pulmonol 2009;44(1):14-30.