Intrabdominal Ewing Sarcoma of the Peritoneum: A Case Report
Abstract
Extraskeletal ewing sarcoma (es) is a small round-cell tumour and constitutes a sparsely reported clinical condition. We present a case of a 19-year-old man with abdominal distension and palpable masses in all quadrants of the abdomen. The patient was diagnosed with peritoneal es and had multimodal oncologic treatment. On neoadjuvant setting, the patient received 9 cycles of chemotherapy and then underwent radical surgical excision, achieving r0 resection. Subsequent to an unremarkable postoperative course, 5 cycles of adjuvant chemotherapy were implemented to achieve optimal survival outcomes. On the current follow up, there is no evidence of local or further systematic spread of the disease. Es is a very rare malignancy and few cases have been published with only intraperitoneal spread. Chemosensitivity of the tumor is characteristic, making possible r0 resections, however local recurrence still exists.
Keywords: extraskeletal ewing sarcoma; extraosseous ewing sarcoma; peritoneum
Introduction
Ewing sarcoma (es) constitutes a malignant small round-cell neoplasm predominantly originating in skeletal and soft tissues, with a stated incidence of 1 case per million1,2. Extraskeletal es is a rare clinical entity comprising 20% of es3. The trunk is the most frequently impacted site, followed by the extremities, head and neck and the retroperitoneum3. We present a rare case of a 19-year-old man diagnosed with excessive es of the peritoneum.
Case
report
A
19-year-old man presented to our department with massive abdominal distension,
persisting for two months. His medical history was unremarkable and upon
clinical examination a distended abdomen and palpable masses were found in all
four quadrants, raising suspicion of sarcoma. Laboratory diagnostic tests were
normal, with the exception of leukocytosis with neutrophil predominance.
Following an ultrasound examination which revealed hypoechoic heterogeneous
masses, a contrast-enhanced computed tomography (ct) scan yielded mild ascites
and significant opacity of the peritoneal fat, along with heterogenous focal
lesions. The largest mass was located in the lesser pelvis, measuring 20 x 14.5
x 16 cm, accompanied by concurrent enhancement in the paracolic gutters and
omentum, indicating further peritoneal dissemination (figure 1a and 1b).
Magnetic resonance imaging (mri) confirmed the ct findings, displaying a mass
with restricted diffusion and central necrosis and established the differential
diagnosis of a primary peritoneal lesion, sarcoma or stromal tumour (figure
2). Ultrasound-guided biopsy and histological examination established the
diagnosis of extraskeletal es and the patient commenced neoadjuvant
chemotherapy. He underwent 9 cycles of chemotherapy every 15 days, consisting
of 4 cycles of ifosfamide, etoposide, mensa d1-d5 (ie) alternating with 5
cycles of cyclophosphamide, vincristine, doxorubicin, mensa (vdc) d1. The
patient exhibited remarkable treatment response on follow-up ct scans, with a
regression of ascitic fluid and a decrease in the size of the mass in the
lesser pelvis to 3.5 x 2.5 cm, as well as of the concomitant lesions in the
paracolic gutters and the omentum (figure 3). Subsequently, the patient
underwent radical surgical excision of the peritoneal disease along with
partial enterectomy 15 cm from the ileocecal valve due to local infiltration,
with the objective of r0 resection. The postoperative recovery was uneventful
and the patients was discharged on postoperative day 7. Histopathological
analysis of the respected specimen confirmed the diagnosis of primary
peritoneal es. Following surgery, the patient received 3 cycles of ifosfamide,
etoposide, mensa d1-d5 (ie) alternating with 2 cycles of cyclophosphamide,
vincristine, doxorubicin, mensa (vdc) d1. On the present follow-up, no evidence
of recurrence has been indicated.
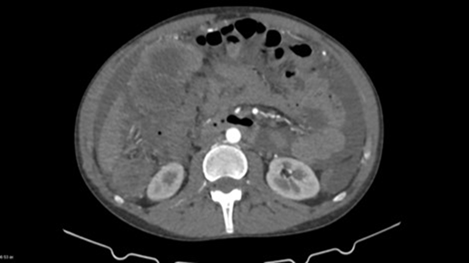
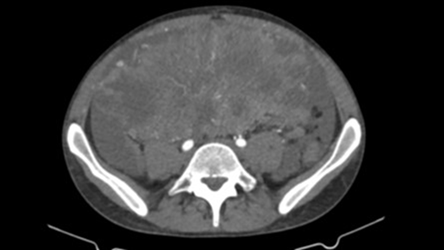
Figure 1a
& 1b: preoperative
contrast enhanced ct scan of the largest lesion measuring 20 x 14.5 x 16 cm.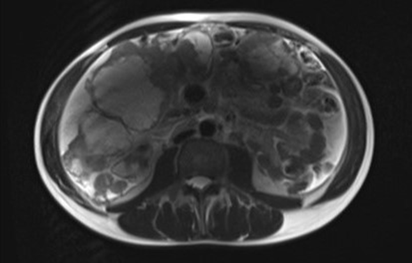
Figure 2: abdominal
mri: the mass has high intensity on t2-weighted images
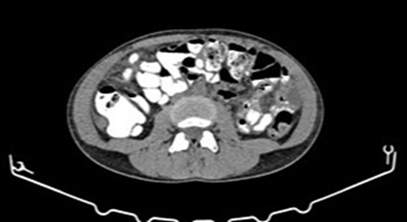
Figure 3: post-chemotherapy contrast enhanced ct scan for
tumour response assessment
Discussion
Ewing
sarcoma arises from a family of highly malignant small round cell tumours,
defined by a shared genetic, histological and immunohistochemical basis4. This spectrum also encompasses extraskeletal
es, small-cell tumour of the thoracopulmonary region (askin tumor) and soft
tissue based primitive neuroectodermal tumours (pnets)4. The predominant chromosomal translocation
among 90% of es family members is t (11;22) (q24; q12), resulting in the
ewsr1-fli1 fusion oncogene1,3.
Although it continues to be a sparsely documented condition in the literature,
extraskeletal es was first described in 19695.
Unlike skeletal es, extraosseous es exhibits a higher mean age of onset and a bimodal age distribution, being more prevalent in patients younger than 5 and older than 35 years old1,3. Extraosseous es is distinguished by its axial tumor origin and rarely exceeds 8 cm in diameter3,6. The clinical image of extraosseous es is non-specific, resulting thus in delay of diagnosis, with a growing mass causing localised pain as the most prevalent manifestation7. Other symptoms vary according on the site of origin and may include fever, weight loss, fatigue and metastasis-derived symptoms7.
While the imaging findings are likewise non-specific, imaging serves as important for the diagnosis, staging and subsequent follow-up of patients8. Ultrasonography demonstrates a hypoechoic mass displaying heterogeneity and intratumoral flow signs on doppler imaging7. Similarly, ct scan indicates a defined mass with density equivalent to or lower than that of the muscle, while mri yields a low to intermediate and a high intensity signal on t1 and t2-weighted images, respectively. Regarding contrast enhancement, extraosseous es exhibits an heterogenous pattern associated with intratumoral necrosis in both imaging modalities3,7. Mri contributes to staging by assessing the local extent of the tumor, whereas fdg-pet/ct serves as the primary staging modality for identifying distant metastases3. Extraskeletal es doesn’t spread through the lymphatic pathways, but rather by hematogenous routes, often localising in lungs, followed by bone and central nervous system involvement8. Metastatic development to other organs of the abdominal cavity, the peritoneum or surrounding soft tissue is uncommon, with a reported rate of 10%3,8. Meanwhile, a study by eary et al illustrated that the standardised uptake value (suv) of the primary tumor in fdg-pet/ct provides predictive information regarding patients’ overall survival9.
The conclusive diagnosis of extraskeletal es necessitates histological analysis in conjunction with immunohistochemistry and genetic examination1. Differential diagnosis may prove challenging and consists of several small round cell neoplasms, including other tumours from es family, embryonal rhabdomyosarcoma, neuroblastoma and lymphoblastic lymphoma3,7.
Despite that optimal treatment protocols remain undetermined for extraskeletal es, national comprehensive cancer network (nccn) suggests for the localised ewing sarcoma family of tumours localised treatment (surgery and/or radiotherapy) alongside chemotherapy1,10. Following neoadjuvant chemotherapy, surgical excision of the primary tumor is recommended aiming for r0 resection, while radiotherapy is implemented in case of positive margins or in inoperable cases1,3. The prognosis of extraosseous es is superior as opposed to the osseous subtype with five-year overall survival rates for localised tumours being 70%, whereas for metastatic disease 33%1,3,7. Metastatic spread represents the fundamental prognostic indicator with pelvic involvement and older age indicating a poor prognosis1. On the contrary, response to the neoadjuvant chemotherapy and radical surgical excisions have been correlated with favourable outcomes1.
Conclusion
The present case report, which presents the peritoneal
infiltration of extraosseous es, emphasises on the challenge of diagnosis
arising from disease entity’s rarity. A high level of clinical suspicion,
alongside clinical, imaging and histopathological findings are essential for
ensuring optimal patient care. Thus, a multidisciplinary team approach,
implementing reductive surgeries combined with chemoradiotherapy, is required
to address this uncommon clinical condition and achieve favourable survival
rates.
Consent for publication
Written informed
consent was obtained from the participant for the publication of the details of
their medical case.
Availability of data and materials
Not applicable.
Conflicts of interest
All authors
declare no potential conflicts of interest with respect to the research,
authorship and/or publication of this article.
Funding
This study was not
supported by any sponsor or funder.
Author contributions
Conceptualization-
d.l, methodology- e.m, data collection and formal analysis- e.m, s.c, k.p
writing-original draft- d.l, e.m, s.c, writing- review and editing- d.l, k.p,
a.m, supervision- a.m. all authors have read and agreed to the published
version of the manuscript.
Acknowledgements
None.
References
1.
Abboud a, masrouha k, saliba m et al. Extraskeletal
ewing sarcoma: diagnosis, management and prognosis. Oncol lett 2021;21(5):354.
2. Jawad mu, cheung mc, min es,
schneiderbauer mm, koniaris lg, scully sp. Ewing sarcoma demonstrates racial
disparities in incidence-related and sex-related differences in outcome: an
analysis of 1631 casesfrom the seer database, 1973-2005. Cancer 2009;115(15):3526-3536.
3. Revannagowda s, gangadhar k, akaike
g, dighe m. Primary intra-abdominal ewing's sarcoma in adults: a multimodality
imaging spectrum. Curr probl diagn radiol
2020;49(2):133-139.
4.
Grier he. The ewing family of
tumors. Ewing's sarcoma and primitive neuroectodermal tumors. Pediatr clin north am 1997;44(4):991-1004.
5.
Tefft m, vawter gf, mitus a.
Paravertebral "round cell" tumors in children. Radiology 1969;92(7):1501-1509.
6. Cash t, mcilvaine e, krailo md et
al. Comparison of clinical features and outcomes in patients with extraskeletal
versus skeletal localized ewing sarcoma: a report from the children's oncology
group. Pediatr blood cancer 2016;63(10):1771-1779.
7.
Galyfos
g, karantzikos ga, kavouras n, sianou a, palogos k, filis k. Extraosseous
ewing sarcoma: diagnosis, prognosis and optimal management. Indian j surg
2016;78(1):49-53.
8. Javery o, krajewski k, o'regan k, kis b, giardino a, jagannathan j, ramaiya nh. A to z of
extraskeletal ewing sarcoma family of tumors in adults: imaging features of
primary disease, metastatic patterns and treatment responses. Ajr am j roentgenol 2011;197(6):1015-1022.
9.
Eary jf, o'sullivan f, powitan y, et al. Sarcoma tumor fdg uptake measured by pet and patient
outcome: a retrospective analysis. Eur j nucl med mol imaging 2002;29(9):1149-1154.
10. Casali pg, bielack s, abecassis n et
al. Bone sarcomas: esmo-paedcan-euracan clinical practice guidelines for
diagnosis, treatment and follow-up. Ann oncol 2018;29(4):79-95.