Lymphomatoid Papulosis in Pediatric Patient: A Case Report
ABSTRACT
Lymphomatoid
papulosis is a low-grade malignant skin lymphoma. It is a rare
lymphoproliferative disease clinically characterized by generalized chronic,
recurrent and self-limited CD30+ positive papules and nodules. It courses along
with a mixed inflammatory infiltrate of eosinophils, neutrophils, histiocytes
and plasma cells that in histopathology can adopt a variety of at least six
patterns named from A to F and it is possible to see more than one variable in
one patient; it can eventually progress to a primary skin lymphoma of large
anaplastic cells.
Keywords:
Lymphomatoid Papulosis; Mycosis fungoides; Lymphoproliferative
disorders; Cutaneous lymphoma.
Abbreviations:
LP:
Lymphomatoid papulosis;
ALCL:
Primary cutaneous anaplastic large cell
lymphoma;
PLEVA:
Pityriasis lichenoides et varioliformis
acuta;
PLC:
Pityriasis lichenoides chronica;
MF;
Mycosis fungoides;
EBV:
Epstein-Barr virus
INTRODUCTION
Lymphomatoid
papulosis (LyP) is a self-regressing, chronic, CD30 T-cell lymphoproliferative
disease characterized by recurrent, and spontaneously remitting papulonodular
or necrotic lesions that appear anywhere on the body but are frequently
disseminated1. It is classified alongside primary
cutaneous anaplastic large cell lymphoma, in the group of T-cell proliferations
expressing CD30.
In
roughly 10–20% of the patients, it is linked to an increased risk of secondary
lymphomas, including mycosis fungoides and CD 30+ large T-cell lymphoma (LTCL).
This disorder's most remarkable aspect is that its aggressive histological
characteristics that closely resemble lymphoma, are not concordant with the
benign clinical course and spontaneous remission2.
Due to the increased risk of developing non-Hodgkin lymphoma, lifelong
follow-up is justified. We report
the case of a 9-year-old presenting with recurring papulo-necrotic lesions over
face, trunk, and extremities.
CASE PRESENTATION
We
received a 9 year-old girl with a history of 18 months of recurrent papular
lesions which progressed slowly and appeared mostly in the limbs, treated with
emollients and topical and systemic steroids without total improvement. The
lesions had changed morphology and generalized. At the physical examination she
presented a dermatosis disseminated to trunk and limbs characterized by
multiple erythematous papules and papulo-vesicles ranging from 3 to 10 mm of
diameter, some ulcerated with hemorrhagic crusts on its surface (Figure 1). she also presented some
very pruritic nodules of approximately 5 mm accompanied by mild xerosis without
signs of over infection. The clinical differential diagnosis included
pityriasis lichenoides chronica versus lymphomatoid papulosis. Cutaneous biopsy
of the right leg was performed revealing an infiltrate extending from the
papillary to the reticular dermis in smaller extent, composed mainly by
lymphocytes, some histiocytes, neutrophils and eosinophils, as well as a
smaller number of large atypical lymphocytes (Figure 2). Immunohistochemistry stained strong CD4+ expression in
approximately 90% in a wedge shape. Additionally, epidermo and folliculotropic
CD30+ cells (Figure 3), with
moderately strong perinuclear and cluster expression (Figure 4) appeard. Normally, CD4+ is dominant in CD30+
lymphoproliferative diseases, thus the diagnosis of Papulosis Lymphomatoid Type
A was made. The patient started methotrexate 10 mg weekly which lead to the
involution of lesions leaving post-inflammatory scars. After 1 year, treatment
was deescalated to tacrolimus 0.1% proactive twice weekly and emollients in
lesions. The patient continues follow up every 3 months.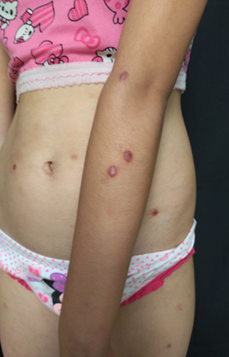
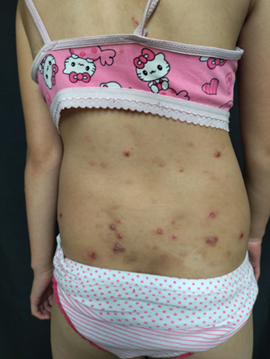
Figure 1.
Multiple erythematous papules and papulo vesicles of several mm of diameter,
some ulcerated, covered with hemorrhagic crusts over trunk and extremities(A, B). 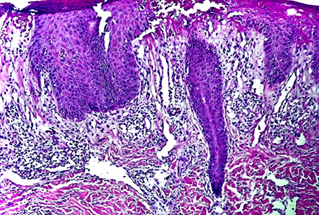
Figure 2.
Infiltrate that extends from the papillary dermis to the reticular dermis in
smaller extent, composed mainly by lymphocytes, histiocytes some neutrophilus
and accompanyed by einophilus, and a smaller number of large atypical
lymphates.
Courtesy Dra.
Johanna Brito - LunaPiel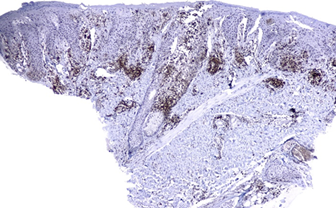
Figure 3. Strong expression CD4+ approximately
90% in a wedge shape, in addition to epidermotropism and folliculotropisms of
cells
Courtesy Dra.
Johanna Brito - LunaPiel 
Figure
4.
Moderately strong perinuclear and cluster CD30+ expression
Courtesy Dra.
Johanna Brito – LunaPiel
Disseminated papulonodular eruptions that spontaneously resolve in a matter of weeks to months are the typical presentation of LyP. Healing lesions manifest as temporary hyper or hypopigmented macules following the resolved inflammation, and they can also occasionally develop into atrophic varioliform scars.
Although it has been proposed that the spontaneous resolution of lesions and the small number of cases diagnosed during pediatric age may underestimate its frequency, LyP is most typically observed in adults. The course and clinical manifestation of LyP in children does not differ much from that of adults. LyP is the second most common cutaneous lymphoproliferative disease after mycosis fungoides, although it is indeed uncommon in pediatric patients3.
The etiology of lymphomatoid papulosis is unknown. Numerous multidisciplinary studies have focused on the etiology and pathogenesis of LyP, including the processes underlying the disease's spontaneous remission. Researchers are searching for evidence of participation in the etiopathogenesis of oncogenic viruses such as Epstein Bar or herpes virus, atopy (seen in around 50% of patients), genetic susceptibility factors (aneuploidy and chromosomal aberrations), and immune system abnormalities4.
Based on histologic features, LyP is divided into five subtypes (A to E), with type A being the traditional and most prevalent type (>75%)5, characterized by a wedge-shaped dense dermal perivascular lymphoid infiltrate with large atypical CD30-positive cells with Reed-Sternberg appearance; Type B has cerebriform cells and epidermotropic lymphocytes similar to MF; Type C resembles anaplastic large cell lymphoma (ALCL), with sheets of CD30+ large cells; type D is characterized by a CD8+ infiltrate mimicking the features of cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma; type E is characterized by eschar-like necrosis, ulceration and larger papules as a consequence of the infiltration and destruction of dermal and subcutaneous vessels; type F that has CD30+ atypical lymphocytic infiltration of the follicular epithelium clinically gives rise to a papular and/or pustular phenotype as neutrophils and eosinophils are attracted to the infiltrate6.
The differential diagnosis of LyP includes arthropod bites, primary cutaneous anaplastic large cell lymphoma (ALCL), a papular variant of mycosis fungoides, and both forms of pityriasis lichenoides (pityriasis lichenoides et varioliformis acuta, PLEVA, and pityriasis lichenoides chronica, PLC)3.
Due to the recurrent and chronic nature of LyP, the treatment is usually symptomatic and aimed at accelerating the resolution of lesions or reducing their severity. Strong topical corticosteroids, intralesional steroids, or surgical excision are effective treatment choices for one to a few isolated papules. Patients with significant or symptomatic disease, or disease affecting cosmetically sensitive areas such as the hands or face, should start with low-dose methotrexate as the first line of treatment. While various signs have been proposed, there is currently no way to forecast how a patient's condition would progress. Patients should be monitored for the remainder of their lives due to the lack of markers that can assist forecast the course of the disease and the occurrence of malignant lymphoma7.
CONCLUSION
Lymphomatoid papulosis is a rare lymphoproliferative disorder in the pediatric and adolescent subpopulation, it is indolent and with a higher rate of spontaneous regression.
This case underlined the importance of diagnostic confirmation with the clinicopathological and immunohistochemical distinction. It is also essential to recognize histologic characteristics of LyP type A to halt misdiagnosis. CD30 is the most important immunohistochemistry marker for diagnosis. Additionally, long-term follow-up and subsequent biopsies should be considered in progressive or recalcitrant cases in order to give a precise diagnosis, provide proper management, and evaluate for associated secondary hematologic malignancies.
Ethical permission: The patient has given informed consent during his treatment for the publication of this article.
Conflict of Interest: The authors declare no conflicts of interest.
REFERENCES
1. Toumi A, Fazal S, Litaiem N. Lymphomatoid Papulosis. StatPearls 2023.
2. Verma D, Lakhani R, Mendiratta V, Chatterjee P. Lymphomatoid papulosis: A case report. Indian Dermatol Online J 2023;15(1):95-98.
3. Gomes N, Nogueira A, Silva R, Azevedo F. Multiple painless papulonodules in a 3-year-old girl: type A lymphomatoid papulosis. An bras dermatol 2022;97(5):689-690.
4. Nowicka D, Mertowska P, Mertowski S, et al. Etiopathogenesis, diagnosis, and treatment strategies for Lymphomatoid Papulosis with particular emphasis on the role of the immune system. Cells 2022;11(22):3697.
5. Pomsoong C, Suchonwanit P, Chanprapaph K, Rattanakaemakorn P, Rutnin S. Pityriasis lichenoides Et varioliformis acuta and lymphomatoid papulosis Type F: A case report of two entities in one patient. Clin Cosmet Investig Dermatol 2022;15:1759-1765.
6. Martinez-Cabriales SA, Walsh S, Sade S. Shear NH. Lymphomatoid papulosis: An update and review. J Eur Acad Dermatol Venereol 2020;34:59-73.
7. Balic A, Bartolic L, Ilic I, Rados J. Long-term Follow-up of a Case of Lymphomatoid Papulosis with a benign course. ADC 2018;26(3):264-266.