Novel Hexokinase 1 Genetic Mutation Presenting with Recurrent Fever and Developmental Delay: Possible Insight on the Role of Glucose Metabolism Dysregulation and Autoinflammation
ABSTRACT
Hexokinase 1(HK1) is a key enzyme in the glycolytic pathway.
Dysregulation of HK1 causes activation of NLRP3 inflammasome and subsequently
overexpression of proinflammatory cytokines IL-1 B and IL-18. HK1 genetic
mutation has been reported in cases of neurodevelopment abnormalities with
visual impairment and nonspherocytic hemolytic anemia, but its role in
recurrent fever and systemic inflammation has not been reported yet. we present a case of an early childhood boy with a history of
developmental delay who experienced recurrent episodes of high-grade fever,
responding partially to antipyretics and antibiotics, with abdominal pain but
no skin rash or joint pain. Whole exome sequencing revealed a novel
heterozygous variant in HK1 (c.2198C>T; p. Ser733Phe), which is predicted to
have a potentially deleterious effect based on PolyPhen-2 and SIFT In silico
analyses. This case report
highlights the possible new association of autoinflammation and HK1 mutation in
addition to typical features such as neurodevelopmental delay and anemia.
Keywords: Hexokinase1; Autoinflammation; Neurodevelopmental ; Anemia;
Recurrent fever
Abbreviations
HK1: Hexokinase 1
NLRP3: nucleotide-binding domain, leucine-rich–containing family,
pyrin domain–containing-3
SIFT: Sorting Intolerant From Tolerant
SAD:
Systemic autoinflammatory diseases
ATP:
adenosine triphosphate
ADP:
adenosine diphosphate
G6P:
glucose-6-phosphate
NAG:
N-acetylglucosamine
CNS:
central nervous system
INTRODUCTION
Systemic
autoinflammatory diseases (SAD), also known as periodic fever syndromes, are a
group of disorders with an increasing prevalence that arise from dysregulation
of the innate immune system1. These disorders typically present
with recurring fevers and inflammation without any obvious trigger1.
Many SAD patients have pathological genetic variants contributing to systemic
inflammation2.
In the last three years, the number of autoinflammatory diseases due to
monogenic defects has increased from 42 disorders to 56 disorders based on the
updated classification by the International Union of Immunological Societies in
20223.
Identifying new genetic defects in SAD patients has allowed for a better
understanding of the underlying immunological mechanisms, thus opening new
perspectives in targeted therapies.
The
activation of the NLRP3 (nucleotide-binding domain, leucine-rich–containing
family, pyrin domain–containing-3) inflammasome plays an essential role in
host defense against microbial infections. However, its dysregulated activation can lead to
several autoinflammatory disorders4.
Hexokinase, a key enzyme in the glycolytic pathway, can be released from
mitochondria and activates NLRP3 upon exposure to N-acetylglucosamine, which is
bacterial-derived peptidoglycan5. The activation of NLRP3
inflammasome activation results in the secretion of IL-1B and IL-18, leading to
an inflammatory response. In this report, we present a case of an early
childhood boy with developmental delay who experienced recurrent episodes of
high-grade fever with abdominal pain in the absence of clear evidence of
infection or malignancy. Whole exome sequencing revealed a novel heterozygous
variant in HK1 (c.2198C>T; p. Ser733Phe).
CASE PRESENTATION
We report the case of an early
childhood boy who was born to healthy non-consanguineous parents via
spontaneous vaginal delivery at full term with a birth weight of 2.0 kg. At
eight months of age, he was admitted to the hospital with a seizure and was diagnosed
with aseptic meningitis. The cerebrospinal fluid analysis showed negative
bacterial culture, leukocyte count of 10, glucose of 4 mmol/L, and protein of
21.0 mg/dL. An abnormal Electroencephalogram with epileptiform brain activity
was also identified. He was discharged home on antiepileptics. Subsequently,
The patient was noted to have delayed developmental milestones. He rolled over
at 12 months of age, sat up at 18 months of age, and walked at the age of 30
months. He started to have recurrent episodes of high-grade fevers at the age
of 1 year. Each episode lasted 4-5 days and responded partially to antipyretics
and oral antibiotics. The interval between fever attacks was irregular,
sometimes only a few days apart. The patient was entirely well between the
episodes of fevers. These fever attacks were associated with abdominal pain. He
had no history of skin rash, joint pain, or symptoms suggestive of respiratory
or urinary infections. There was no family history of periodic fevers,
arthritis, or renal failure. Physical assessment at the age of 4 years revealed
a well-appearing non-dysmorphic child. His temperature was 38.8°C, and other
vital signs were normal. His weight was 14 kg (25th centile),
height 98 cm (25th centile), and head circumference 50 cm (50th
centile). A delay in fine and gross motor development was noted. There was no
rash, lymphadenopathy, or other significant findings on physical examination.
Complete blood count revealed normal
white blood cell counts, normochromic microcytic anemia (hemoglobin 10.2 g/dl),
and thrombocytosis )platelet counts 525 X 103/UI). The platelet
count returned to normal between attacks. Iron profile was low and peripheral
blood smear suggested iron deficiency anemia. Liver and renal function tests
were normal. Erythrocytes sedimentation rate was 83mm/h and remained high
between attacks. C-reactive protein was 3.0mg/dl. Screening for infectious
causes was negative and Immunoglobulins levels were normal. Eyes examination by
slit lamp was normal. Abdominal ultrasonography showed mesenteric lymphadenitis
and mild ascites. Echocardiogram was normal. The hearing assessment was normal.
Upon susception of periodic fever syndromes, a commercially available whole
exome sequence was performed. The results revealed a novel heterozygous variant
in Exon/ Intron 15 of HK1 (c.2198C>T; p. Ser733Phe), which is predicted to
have a potentially deleterious effect based on PolyPhen-2 and SIFT In silico
analyses.
DISCUSSION
Our patient was diagnosed with HK 1
genetic mutation by genetic testing, ordered in the setting of recurrent fever,
development delay, and family history of consanguinity. HK1 is a crucial
glucose metabolism enzyme located on the mitochondrial cell wall. It
phosphorylates hexoses (six-carbon sugar) to hexose 6 phosphate by utilizing a
phosphate from the conversion of adenosine triphosphate (ATP) to adenosine
diphosphate (ADP) (Figure 1).
Glucose is the primary substrate for HK1 and glucose-6-phosphate
(G6P) is the major product (Figure1). 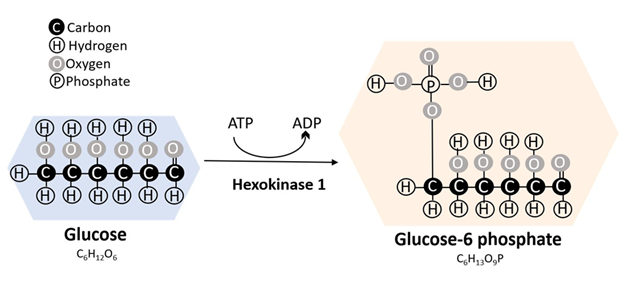
Figure 1: Hexokinases are crucial enzymes involved in glucose metabolism.
They phosphorylate hexoses (six-carbon sugar) to hexose 6 phosphate by
utilizing a phosphate from the conversion of adenosine triphosphate (ATP) to
adenosine diphosphate (ADP).
Phosphorylation of glucose to G6P by
HK1 facilitates glucose transport into cells6. Four major HK isozymes have been
described in humans, each encoded by a distinct gene and expressed in different
tissues6. HK1 is expressed in multiple normal tissues, including
erythrocytes, brain, and fibroblasts, as well as in abnormal cells such as
malignant tissues7,8. HK1 is also expressed in white
blood cells, particularly monocytes, and plays a significant role in glucose
metabolism, which serves as a crucial source of energy for monocytes and
neutrophils9,10. Besides its involvement in energy
utilization and the survival of mononuclear cells, HK1 also plays a vital role
in cell signaling and protection against pathogens5,9. Dysregulation in HK1 enzyme
activity affects these cells leading to the activation of the NLRP3
inflammasome5 (Figure 2).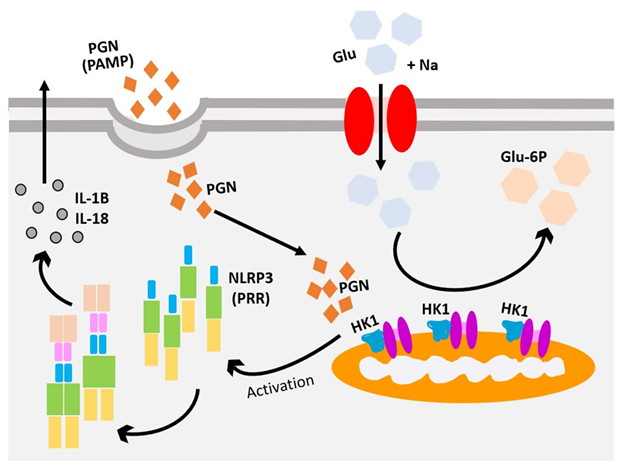
Figure 2: Hexokinase 1 (HK1) binds to
N-acetylglucosamine (NAG) and dissociates from the mitochondria. The
dissociation of HK1 from mitochondria is enough to induce the activation of the
NLRP3 inflammasome and the production of IL-1β.
NLRP3, a high-molecular-weight
protein located on the long arm of chromosome 111, is a Pattern Recognition Receptor
that recognizes Pathogen Associated Molecular Patterns12. NLRP3 gain of function mutation
leads to the autoactivation of the inflammasome pathway and subsequent
secretion of IL-1, resulting in cryopyrin-associated periodic fever syndromes11. Anakinra (Kineret), rilonacept
(Arcalyst), and canakinumab (Ilaris) are medications that inhibit the activity
of IL-1β and lead to the improvement of symptoms and a decrease in systemic
inflammation13.
HK1 has the ability to function as
an innate immune sensor by binding to N-acetylglucosamine (NAG), a
peptidoglycan subunit derived from a gram-positive bacterial cell wall14. The binding of NAG to HK1 inhibits
its activity and leads to the dissociation of HK1 from mitochondria5. The dissociation of HK1 from
mitochondria is enough to induce the activation of the NLRP3 inflammasome and
the production of IL-1β5. It is plausible that the novel HK1
mutation (c.2198C>T; p.Ser733Phe) in our patient results in the spontaneous
dissociation of HK1 from mitochondria, potentially triggering inflammation.
However, further experimental studies are required to confirm this hypothesis.
Traditionally,
HK1 deficiency has been associated with autosomal recessive
early-onset severe nonspherocytic hemolytic anemia caused by an impaired
glycolytic pathway in red cell metabolism15,16. However, additional phenotypes
have emerged over time, including Hereditary Motor and Sensory Neuropathy-Russe17, retinitis pigmentosa18, and neurodevelopmental disorder19. In cases where patients exhibited
neurodevelopmental abnormalities and visual impairment, HK1 mutations were
inherited in an autosomal dominant manner and were not accompanied by hemolytic
anemia. Furthermore, the activity of HK1 in the red blood cells of two patients
was found to be normal, indicating that the neurological manifestations were
not due to a loss of hexokinase enzymatic activity19. Similarly, Sullivan et al.
identified a large six-generation family with autosomal dominant retinitis
pigmentosa caused by a heterozygous HK1 mutation (p.Glu847Lys) without evidence
of hemolytic anemia18. Our patient had a novel HK1
mutation (c.2198C>T; p.Ser733Phe) which presented with recurrent fever and
neurological features but no evidence of hemolysis. While the neurological
manifestations in this case are likely attributed to the direct metabolic
effects of HK deficiency, it is plausible that some features may be influenced
by an inflammatory response in the brain. For example, certain autoinflammatory
disorders are well recognized for their central nervous system (CNS)
involvement20, and other diseases had CNS
features as a recent expansion in their phenotypic presentation21. Additionally, CNS involvement can
be only sign of autoinflammatory disorder on presentation22 as the sole initial sign of an
autoinflammatory condition, CNS involvement can be the only sign of
autoinflammatory condition initially. Lastly, a growing body of evidence
supports the role of neuroinflammation in the development of neurodegenerative
diseases such as Alzheimer's and Parkinson's disease23.
CONCLUSION
In summary, our patient exhibited an
autoinflammatory phenotype characterized by recurrent fever and elevated
inflammatory markers in the absence of an infectious trigger. This expands the
disease phenotype associated with HK1 mutations. The autoinflammatory aspect of
the disease may be unique to this novel mutation (c.2198C>T; p.Ser733Phe)
and its impact on NLRP3 signaling, although further validation is required
through experimental animal models. This case highlights the importance of
genetic testing in patients presenting with neurological sequelae and recurrent
fever, as it can provide crucial insights into the underlying genetic basis of
the condition.
DECLARATIONS
Ethics approval and consent to
participate: ethical approval was obtained from the biomedical research ethics committee at Umm Al-Qura University (Approval No: HAPO-02-K-012-2024-02-2063). Written informed consent was obtained
from the parent.
Authors' contributions: Aisha Mirza wrote the initial draft and
created the figures. Husni Rayes reviewed and edited the manuscript. Heba
AlQurashi reviewed and edited the manuscript. Amer Khojah wrote the conclusion
and edited the manuscript.
Acknowledgements: None
Consent for publication: Consent for publication was obtained from the parent.
Availability of data and material: Data and material will be available from
the corresponding author upon reasonable request.
Competing interests: The authors declare that they have no conflicts of interest related to
this research.
Funding:
None
REFERENCES
1. Krainer
J, Siebenhandl S, Weinhausel A. Systemic autoinflammatory diseases. J Autoimmun 2020;109:102421.
2. Demir F, Dogan OA,
Demirkol YK, et al. Genetic panel screening in patients with clinically
unclassified systemic autoinflammatory diseases. Clin Rheumatol
2020;39(12):3733-3745.
3. Tangye
SG, Al-Herz W, Bousfiha A, et al. Human Inborn Errors of Immunity: 2022 Update
on the classification from the international union of immunological societies
expert committee. J Clin Immunol 2022;42(7):1473-1507.
4. Blevins HM, Xu Y, Biby S,
Zhang S. The NLRP3 inflammasome pathway: A review of mechanisms and inhibitors
for the treatment of inflammatory diseases. Front Aging Neurosci
2022;14:879021.
5. Wolf AJ, Reyes CN, Liang
W, et al. Hexokinase is an innate immune receptor for the detection of
bacterial peptidoglycan. Cell 2016;166(3):624-636.
6. Kanno
H. Hexokinase: Gene
structure and mutations. Best Practice Research Clinical Haematology
2000;13(1):83-88.
7. Shinohara Y.
[Identification and characterization of hexokinase isozyme predominantly
expressed in malignant tumor cells]. Yakugaku Zasshi 2000;120(8):657-666.
8. Wilson
JE. Isozymes of mammalian hexokinase: Structure,
subcellular localization and metabolic function. J Exp Biol 2003;206(12):2049-2057.
9. Sen S, Kaminiski R,
Deshmane S, et al. Role of hexokinase-1 in the survival of HIV-1-infected
macrophages. Cell Cycle 2015;14(7):980-989.
10. Pithon-Curi TC, De Melo
MP, Curi R. Glucose and glutamine utilization by rat lymphocytes, monocytes and
neutrophils in culture: a comparative study. Cell Biochem Funct 2004;22(5):321-326.
11. Hoffman HM, Wright FA,
Broide DH, Wanderer AA, Kolodner RD. Identification of a locus on chromosome
1q44 for familial cold urticaria. Am J Hum Genet 2000;66(5):1693-1698.
12. Martinon F. Detection of
immune danger signals by NALP3. J Leukoc Biol 2008;83(3):507-511.
13. Romano M, Arici ZS,
Piskin D, et al. The 2021 EULAR/American College of Rheumatology points to
consider for diagnosis, management and monitoring of the interleukin-1 mediated
autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis
factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and
deficiency of the interleukin-1 receptor antagonist. Ann Rheum Dis
2022;81(7):907-921.
14. O'Sullivan
D, Kelly B, Pearce EL. When Hexokinase Gets that NAG-ing Feeling. Cell Metab
2016;24(2):198-200.
15. Bianchi
M, Magnani M. Hexokinase mutations that produce nonspherocytic hemolytic
anemia. Blood Cells Mol Dis 1995;21(1):2-8.
16. van
Wijk R, Rijksen G, Huizinga EG, Nieuwenhuis HK, van Solinge WW. HK Utrecht: Missense mutation in the active site
of human hexokinase associated with hexokinase deficiency and severe nonspherocytic
hemolytic anemia. Blood 2003;101(1):345-347.
17. Hantke
J, Chandler D, King R, et al. A mutation in an alternative untranslated exon of
hexokinase 1 associated with Hereditary Motor and Sensory Neuropathy-Russe (HMSNR). Eur J Human Genetics
2009;17(12):1606-1614.
18. Sullivan
LS, Koboldt DC, Bowne SJ, et al. A dominant mutation in hexokinase 1 (HK1)
causes retinitis pigmentosa. Invest Ophthalmol Vis Sci 2014;55(11):7147-7158.
19. Okur
V, Cho MT, van Wijk R, et al. De novo variants in HK1 associated with
neurodevelopmental abnormalities and visual impairment. Eur J Hum Genet
2019;27(7):1081-1089.
20. Uccelli
A, Gattorno M. Neurological manifestations in autoinflammatory diseases. Clin
Exp Rheumatol 2018;36 (1):61-67.
21. Khojah
A, Gunderman L, Bukhari A, Schutt M, Cohran V. Early-onset Crohn's disease, IgA
nephropathy, and hemophagocytic lymphohistiocytosis in a patient with IL-10
receptor deficiency. Clin Immunol
Communications 2022;2:145-148.
22. Blincoe
A, Heeg M, Campbell PK, et al. Neuroinflammatory Disease as an Isolated
Manifestation of Hemophagocytic Lymphohistiocytosis. J Clin Immunol
2020;40(6):901-916.
23. Kwon
HS, Koh S-H. Neuroinflammation in neurodegenerative disorders: the roles of
microglia and astrocytes. Translational Neurodegeneration 2020;9(1):42.