Persistent Massive Splenomegaly: Gaucher Disease Masquerading as Visceral Leishmaniasis
ABSTRACT
Gaucher Disease is an autosomal
recessive lysosomal storage disease characterized by glucosylceramide
deposition in cells of the macrophage-monocyte system due to deficiency of the
activity of the lysosomal hydrolase β-glucocerebrosidase. We present a case of a
4.5-year-old boy initially diagnosed with visceral leishmaniasis. Diagnosis of
Gaucher Disease type I was considered at a later stage, based on persistent
massive splenomegaly, occasional bone pain and demonstration of bone
involvement by magnetic resonance imaging. Diagnosis of Gaucher Disease was
established following enzyme and genetic testing. A novel mutation of GBA1
gene has been identified in our case. We discuss the unusual co-occurrence of
Gaucher disease and visceral leishmaniasis and highlight the importance of
early diagnosis of GD in cases of excessive splenomegaly when other causes have
been excluded.
Keywords: Gaucher Disease, Visceral Leishmaniasis, Child; GBA1 gene
Gaucher Disease (GD) is the
most common lysosomal storage disorder with an incidence estimated at around
1/40,000 to 1/60,000 births 1 that can reach 1/800 births in
high-risk populations of certain origins1,2.
We present a case of a patient with GD in whom the diagnosis was challenging
because massive splenomegaly was initially attributed to Visceral Leishmaniasis
(VL). Consent has been received from the family.
CASE PRESENTATION
A 4.5-year-old boy was referred
to the Paediatric Emergency Department due to splenomegaly, fatigue and
low-grade fever for 2 days. The boy was born to non-consanguineous parents,
following a full-term normal pregnancy, was fully vaccinated for his age, and
lives in a rural area. No history of traveling was reported. Personal medical
history included surgery for cryptorchidism and was otherwise unremarkable.
Family history included heterozygosity for beta-thalassemia in the paternal
family.
The child’s physical
development was borderline normal, with weight and height at the lowest
percentiles for age. Physical examination revealed pallor, a palpable
non-tender spleen of 8 cm below the left costal margin and a 5 cm palpable
liver below the right costal margin. Laboratory investigations showed
pancytopenia with leucopenia (WBC 3200/μl), lymphocytopenia (700/μl), anemia (Hb 10 g/dl), thrombocytopenia (PLT 104000/μl) with normal renal and hepatic function. Peripheral
blood PCR was positive for Leishmania
infantum and treatment with liposomal amphotericin-B was initiated. The
fever subsided but anemia and excessive splenomegaly persisted (Figure 1). Mantoux, viral markers, blood and urine cultures were negative.
Chest x-ray, hemoglobin electrophoresis and coagulation assays were also found
to be normal.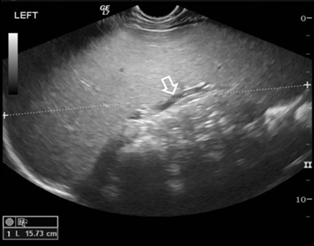
Figure 1: Abdominal
Ultrasound scan. Longitudinal image through the left hypochondrium shows
excessive splenomegaly. The spleen measures 15.7 cm (between cursors) with no
focal lesions and normal diameter of splenic vein (arrow)
Subsequent bone marrow
microscopic examination revealed amastigotes of Leishmania and PCR was found positive for Leishmania 40 days following initial
diagnosis. Two more courses of liposomal amphotericin B and a course of
glucantime and fluconazole were administered for supposed resistant
leishmaniasis in the context of a possible immune defect affecting
intracellular pathogens’ killing, Analysis of lymphocyte subpopulations in
peripheral blood, IL-2/INF-γ axis, vaccine-induced
antibody titers, flow cytometry and karyotype of bone marrow cells
were all normal.
The patient remained afebrile
with occasional bone pain. All serological tests for anti-leishmania antibodies
and subsequent peripheral blood PCR were negative for Leishmania.
Elevated lyso-GL-1 (309.7,
cut-off value <14.0 ng/mL), elevated plasma chitotriosidase activity (4476,
normal value 0-150 nmoles/ml/h) and low β-glucocerebrosidase activity in leukocytes (1.3,
normal value 6-23 nmoles/mg protein/h) were supportive of Gaucher disease. An
abdominal MRI confirmed isolated excessive splenomegaly without significant
intra-abdominal lymphadenopathy (Figure
2). Lower femoral MRI revealed abnormal bone marrow signal for age with
mild Erlenmeyer deformity, suggestive of Gaucher disease (Figure 3).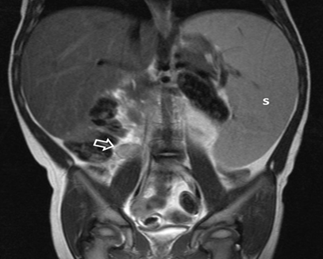
Figure 2: Abdominal MRI scan, Tw-w sequence, coronal plane. The
spleen (S) is excessively large, extending to the left lower abdomen. There is
also isolated mild lymphadenopathy (arrow) at the upper limits of normal for
this location (1 cm short axis).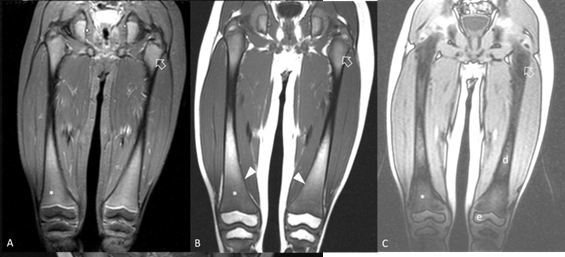
Figure 3:
MRI scan of
femurs, coronal plane. A. STIR sequence, shows intermediate signal intensity at
proximal (arrow) and distal (*) femoral metaphyses which would be expected only
for proximal metaphyses at this age. B. T1-w sequence shows areas of
hypointense bone marrow. There is minimal widening of distal femoral metaphyses
consistent of Erlenmeyer flask deformity (arrowheads). C. Chemical shift
opposed-phase image, confirms signal drop at areas of normal red bone marrow
(arrow) and lack of signal drop at infiltrated metaphyses (*) and at areas of
fatty marrow at epiphyses (e) and diaphyses (d).
Genetic testing by PCR followed
by automated sequencing confirmed the diagnosis of Gaucher disease type 1. Two
mutations in the GBA1 gene were
detected: N370S, already described as
pathogenic for GD, and D283N, a novel mutation not previously identified. The
pathogenicity of the latter was confirmed by in silico tools. Family members’ molecular analysis revealed
heterozygosity for the D283N allele in the father and for the N370S allele in
the mother. Enzyme replacement therapy with imiglucerase was initiated.
The boy’s clinical condition
gradually improved with significant decrease in the spleen size, absence of
bone pain and hemoglobin levels at the lower normal percentiles for his age.
DISCUSSION
Gaucher
Disease is a rare autosomal recessive disorder and it is caused by mutations in
the GBA1 gene, located on the long arm of chromosome 1 (1q21), resulting
in deficiency of the lysosomal hydrolase β-glucocerebrosidase activity1. More than 500 mutations have been described
in GD3. One of the 2 mutations in our
patient, the D283N, is a novel one, while the N370S mutation is a frequent one.
In a recent 35-year cohort study the estimated incidence of GD in Greece is
2.8/100,000 births, and N370S was one of the most frequently identified
mutations accounting for 49.2% of the alleles3.
This mutation is considered neuroprotective and is found only in patients with
type 1 GD3. The D283N mutation has
not been previously described and is one of the 7 novel mutations in the cohort
of GD patients diagnosed in Greece3.
Its pathogenicity, along with other novel mutations according to the same
study, was evaluated with different, well-established predictive tools
(Polyphen2, SIFT and Mutation Taster) and was classified as disease-causing3.
The clinical
manifestations of GD are highly variable. Phenotypic diversity has been
described even in the same genotypic group. GD is subdivided into three
phenotypes. GD type 1 is the most prevalent subtype in the Western world
accounting for ∼90%
of patients worldwide, 85.1% in the Greek cohort3,4.
Our case is a
rare occurrence of GD masquerading as visceral leishmaniasis (VL). Mediterranean basin remains an endemic area for Leishmania Donovani infantum and an
increasing incidence of VL has been reported in Crete5. To our knowledge, two additional cases of VL
associated to GD have been published6,7.
Whether infection by Leishmania may trigger the clinical manifestation of GD or
co-occurrence is a coincidence cannot be defined. This possible new
association supports the need for further studies on GD pathophysiology.
Imaging is
important in supporting the diagnosis and for the follow-up of children with GD8. Ultrasonography and abdominal MRI are the
preferred methods. Abnormal bone marrow involvement in MRI in GD is a prominent
finding, almost uniformly noted in type 1 patients, involving the spine,
femurs, tibias, and less commonly the humeri8.
We
report this unusual case to raise awareness of the possibility of co-existence
of GD and VL in the pediatric population, especially in leishmaniasis endemic
areas where unusual presentations can occur. Diagnosis of GD was confirmed with genetic testing
that revealed a novel, disease-causing mutation of the GBA1 gene. GD should be suspected in persisting massive splenomegaly,
especially in combination with hematological abnormalities and bone pain given
that other causes are excluded. Imaging has its role in the diagnosis of GD.
The availability of a lifelong treatment with favorable outcome in type 1
Gaucher disease mandates early recognition and appropriate treatment.
Summary-Learning points
· The
diagnosis of visceral leishmaniasis especially in endemic areas must not
prevent from testing for Gaucher Disease in the appropriate clinical setting.
· Imaging
has its role in the diagnosis of Gaucher Disease
Acknowledgments
The
authors of the manuscript wish to thank Professor Daniel Grinberg, Department
of Genetics, Microbiology and Statistics, University of Barcelona, for his
contribution to the diagnosis of this case by mutational analysis.
Funding:
None
Conflict of interest:
The authors do not have any conflict of interest to declare.
References
2. Grabowski GA. Gaucher disease: Gene frequencies and genotype/phenotype correlations. Genet Test 1997;1(1):5-12.
3. Dimitriou E, Moraitou M, Cozar M, et al. Gaucher disease: Biochemical and molecular findings in 141 patients diagnosed in Greece. Mol Genet Metab Rep 2020;24:100614.
4. Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: Demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med 2000;160:2835-2843.
5. Antoniou M, Messaritakis I, Christodoulou V, et al. Increasing incidence of zoonotic visceral leishmaniasis in Crete, Greece. Emerg Infect Dis 2009;15:932-934.
6. Gupta SS, Mondal P, Basu N, Mallick M. Gaucher′s disease with uncommon presentations. J Cytol 2009;26:117-119.
7. Zoulati G, Maiga R, Elkhiyat M, Amrani M. Visceral Leishmaniasis and Gaucher disease: Case report of an unusual association. Ann Case Rep 2020;5:520.
8. Degnan AJ, Ho-Fung VM, Ahrens-Nicklas RC, et al. Imaging of non-neuronopathic Gaucher disease: Recent advances in quantitative imaging and comprehensive assessment of disease involvement. Insights Imaging 2019;10(1):70.