Primary Recurrent Stromal sarcoma of Breast: A Rare Case
Keywords: recurrent stromal sarcoma; histopathology; radiotherapy
1. Introduction
Sarcomas of breast are extremely rare breast tumours with an incidence of <1% of all the primary malignancies of breast and < 5% of all the sarcomas1. Estimated annual incidence is 17 new cases per 1,000,000 women2. They arise from mesenchymal tissues of the breast. A 90-year search of the mayo clinic database revealed that primary breast sarcoma accounted for 0.06% of all breast cancers3. Tumour size was the most valuable prognostic factor, with 91% overall survival for women with sarcomas less than or equal to 5 cm and 50% for those with sarcomas greater than 5 cm and unlike carcinoma of any type of mammary sarcoma rarely metastasizes to lymph nodes4.
Poland
et al conducted a study in which, during a period of 80 years, only 4 cases of
fibrosarcoma were reported of the 25 cases of primary breast sarcoma5. Terrier et al, reviewed of 33 cases of breast
sarcoma of which only 2 cases of fibrosarcoma were reported6. Blanchard et al, reported only 2 cases of
fibrosarcoma from his study consisting of 55 sarcoma cases7.
Thorough
tissue sampling is required to exclude a sarcoma arising in malignant phyllodes
tumour, and immunohistology is necessary to exclude a high-grade sarcoma
showing specific mesenchymal differentiation8.
The
most common presentation of fibrosarcoma of breast is rapidly progressing
painless lump in breast which attains a large size. Not much literature is
available on breast sarcomas, due to its rarity in incidence and reporting.
Occurrence
of breast sarcoma is more common in younger age group of 30-40 years age.
Treatment is simple mastectomy with negative margins followed by radiotherapy.
The prognosis is dependent on staging and the histological type of the primary
sarcoma9.
2. Case presentation
A
35year old lady, came to the surgical opd with a complaint of a huge left
breast lump, gradually increasing in size over 10 years, but she noticed rapid
increase in the size seen over the past few months. The lump was painless, not
associated with nipple discharge. There was no history of any hormonal therapy,
radiation exposure or trauma. Married since15 years, breastfed both children
till 2 years; menstrual & obstetrics history were within normal limits.
There were no positive findings in the past, personal or family history either.
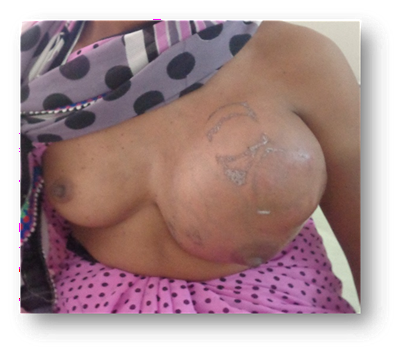
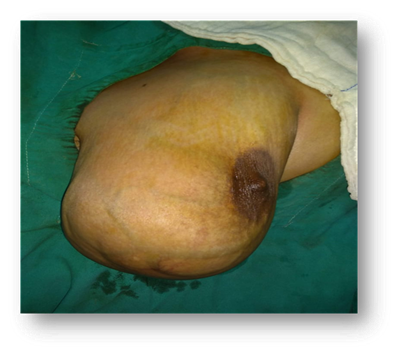
Figure 1. (a) & (b) clinical photo of left breast lump
Clinically (figure 1), there was single, large lump involving the entire left
breast of 22x16cm dimensions, hard in consistency with ill-defined margins and
bosselated surface. Left nipple- areolar complex was at a lower level compared
to the right due to the lump, no nipple discharge seen. The lump was mobile;
not fixed to the underlying chest wall, skin over the lump was pinchable,
dilated veins visible, no dimpling or puckering seen. On investigating further,
the sonomammography reported ‘a large ill defined heterogenous predominantly
hypoechoic lesion note involving the entire left breast parenchyma with flecks
of calcification within and showing vascularity on colour doppler s/o
neoplastic aetiology birads 5 with bilateral axillary lymphadenopathy.’ trucut
biopsy revealed ‘left breast stromal tumour suggestive of phyllodes’. Hence, a
simple mastectomy was done, (figure 2)
post which the histopathology reported ‘left breast stromal sarcoma.
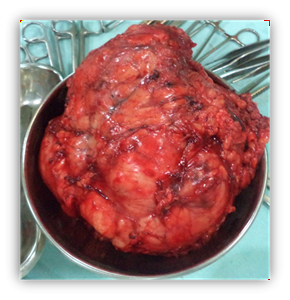
Figure 2. Mastectomy specimen.
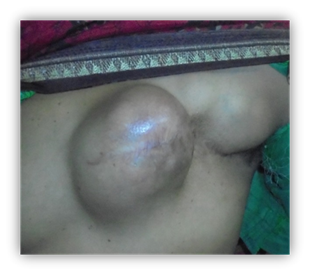
figure 3. Recurrent chest wall tumour.
The patient did not follow up for further treatment advised by the oncologist, thus landing up with a local recurrence, left chest wall and axillary tumour (figure 3). Two lumps 9x8cm over the left chest wall and 3x3 cm in left axilla, both hard in consistency, with irregular surface and rounded margins, mobile, not fixed to the underlying structures, skin above was shiny, not pinchable with previous mastectomy scar visible. Ct thorax reported ‘a well-defined, round lobulated soft tissue lesion 7x4.6x8.6cm over the anterior chest wall, underlying muscle and ribs appear normal however, the lesion has infiltrated into the subcutaneous plane. Similar small lesion 1.6x1.2x1.7cm noted medial to the previous lesion’. A wide local excision with left latissimus dorsi, pedicled rotational flap was done as a definitive surgical treatment.
1.
Histopathology
Gross examination showed tumour near the
base of the specimen with skin infiltration, four separate masses identified,
no lymph node identified. Microscopic examination of the mass showed
interlacing fascicles of spindle shaped cells with spindle shaped nuclei with
tapering cytoplasmic processes. Frequent mitotic activity is noted
(>10/10hpf) suggestive of left breast ‘stromal sarcoma.’ ihc was negative
for cytokeratin and ema. The patient was further referred to radiation
oncologist whereby she received 50gy/25# in total and follows up regularly, no
recurrence or new lesion found elsewhere (figures
5,6).
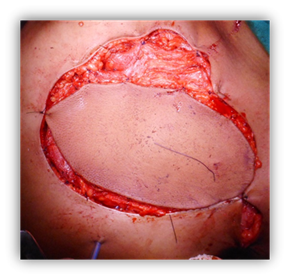
figure 5. Left latissimus dorsi flap to sarcoma of the breast.
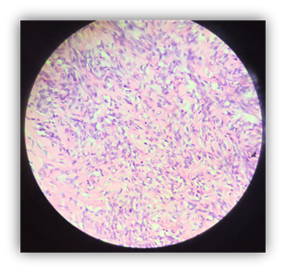
figure 6. Microscopy of stromal cover the defect.
1.
Discussion
Primary stromal sarcoma of the breast
can be of various types like, fibrosarcoma, malignant fibrous histiocytoma,
liposarcoma, rhabdomyosarcoma, leiomyosarcoma, hemangiosarcoma, osteogenic
sarcoma, chondrosarcoma and malignant schwannoma9.
Fibrosarcoma usually occur primarily in the extremities, rarely does it occur
in the breast. The size of primary breast sarcomas is variable and it ranges
from less than 1 cm to larger than 40 cm10.
Primary breast sarcomas may metastasize by hematogenous metastasis or direct
invasion. Rarely axillary lymph node involvement is seen1. And the incidence of actual node metastasis
also appears to be very low11. The
prognosis is dependent on the tumour size and the histopathological grade3.
Terrier et al6. Reviewed 33 cases of primary breast sarcoma
retrospectively and assessment of primary breast sarcoma prognostic factors was
done. Of the total, 17 cases were cysto-sarcoma phyllodes and stromal sarcomas
were 16. The classification of stromal sarco-mas were done as follows-malignant
fibrous histiocytomas (11 cases), leiomyosarcomas (2 cases), fibrosarcoma (2
cases) and liposarcoma (1 case).
In that study, only the histological
grade, consisting of the presence of tumour necrosis, tumour differentiation,
and the mitotic activity was significantly correlated with the metastasis-free
survival rate. Both cysto-sarcoma and stromal groups had identical clinical
courses and survival, thus making the clinical value of this pathologic
distinction questionable. All the local recurrence, metastasis or death
occurred within 30 months, although the follow-up was much longer. Performing
immunohistochemistry has not been very beneficial for identifying the specific
histologic sub-types.
Post-operative radiotherapy helps to
prevent local recurrence. Adjuvant chemotherapy is of some help in patients
with highly malignant sarcoma, with positive surgical margins or post-operative
recurrence12, although the role of
chemotherapy for breast sarcomas remains still unclear. The most effective
regimen for chemotherapy is adriamycin (adm) + ifosfamide (ifo).
2.
Conclusion
Primary breast stromal sarcomas are rare soft tissue tumours diagnosed mainly on histopathology. Surgical management is effective, followed by radiotherapy to avoid local recurrence and chemotherapy in cases of highly malignant tumours, thus demanding a multidisciplinary team approach for the betterment of the patients and efficient management.
3.
Conflict of interest
None declared.
References
1. Moore mp, kinne dw. Breast
sarcoma. Surg clin north am 1996;76(2):383-392.
4. Bland ki, copeland
em. The breast, fourth edition, canada: saunders elsevier 1991;261-264.
8. Bailey h, love m.
Short practice of surgery, 26th edition, florida: crc press 2013; 819.
10. Mcdivitt r, stewart
fw, berg jw. Tumours of the breast. In: atlas of tumour pathology. Washington,
d.c.: armed forces institute of pathology; 1968;127-130. Cited from trent ii jc
2nd, benjamin rs, valero v. Primary soft tissue sarcoma of the breast. Curr
treat options oncol 2001;2:169-176.
11. Roberson gv. Fibrosarcoma of
the breast. J ark med soc 1973;69(9):257-265.