Renal Failure, Spina Bifida, and Porphyria: A Case Report and Review of the Literature
Abstract
This case report details the complex clinical
presentation of a 33-year-old female with a history of meningomyelocele spina
bifida, who developed early-onset renal failure and was later diagnosed with
porphyria. The patient presented with bullous lesions on sun-exposed areas,
which led to the investigation of potential underlying metabolic disorders.
Despite surgical interventions for spina bifida, the patient’s urinary function
remained compromised, eventually progressing to stage 5 vesicourethral reflux and
requiring long-term dialysis. The porphyria diagnosis was confirmed through a
spot urine porphobilinogen test followed by a 24-hour urine analysis, and the
specific subtype was found to be vp (variegate porphyria) confirmed by a
genetic analysis. This case emphasizes the importance of considering porphyria
in patients with unexplained dermatological and neurological symptoms,
particularly when coexisting with other congenital or chronic conditions. The
overlap of renal failure, spina bifida, and porphyria in this patient
underscores the challenges in diagnosis and management, highlighting the need
for multidisciplinary care. Early diagnosis and intervention are crucial to
prevent complications and improve quality of life, particularly in rare and
complex presentations such as this one. This report aims to raise awareness
among clinicians about the potential for delayed diagnosis of porphyria, which
can lead to severe and irreversible complications.
Keywords: porphyria;
dialysis; renal failure; spina bifida; bullous lesions; metabolic disorders
Introduction
Porphyrias
are a group of metabolic disorders caused by enzyme deficiencies in the heme
biosynthetic pathway, often leading to the accumulation of porphyrins and their
precursors. Clinical manifestations vary widely, ranging from cutaneous
symptoms to potentially life-threatening acute neurovisceral attacks. Early
recognition is crucial for preventing complications, yet diagnosis can be
challenging due to the rarity and diverse clinical presentations of these
disorders.
Case presentation
A
33-year-old female was referred to our clinic with recurrent bullous lesions on
her face, arms, and hands following sunlight exposure. These lesions, which
initially left black scars and progressed to permanent white scars, prompted
investigation into underlying metabolic disorders. Her medical history was
significant for meningomyelocele spina bifida at birth, complicating urinary
function despite surgical intervention.
Renal
complications emerged in her early twenties, progressing to stage 5
vesicourethral reflux requiring weekly dialysis. Over eight years, dialysis
frequency increased to three times weekly, managed at home since the age of 28.
Family history was notable for congenital adrenal hyperplasia in her mother and
diabetes mellitus in her father, although no direct familial link to her
condition was identified.
1.
Symptomatic management of porphyria exacerbations:
initiating heme therapy, such as intravenous hemarginate, to alleviate acute
symptoms and prevent porphyria attacks.
2.
Renal supportive care: continuation of
thrice-weekly dialysis, which the patient performs independently at home, to
manage uremic symptoms and maintain fluid and electrolyte balance.
3.
Skin protection strategies: implementing stringent
sun protection measures, including the use of broad- spectrum sunscreen,
protective clothing, and avoidance of direct sunlight exposure during peak
hours, to mitigate cutaneous symptoms associated with porphyria.
In
the treatment regimen for this patient, we administered intravenous hemarginate
at a dose of 3 mg/kg during acute porphyria attacks, which were confirmed by
the ehrlich aldehyde test (figure 1).
This approach was effective in managing the acute symptoms and preventing
further exacerbations. Additionally, we prescribed plaquenil®
(hydroxychloroquine) at a dosage of 100 mg per week to address the chronic
cutaneous manifestations. After a 10-month course of plaquenil®, the
patient's bullous lesions have completely resolved, indicating a successful
outcome in the management of her porphyria-related skin symptoms. 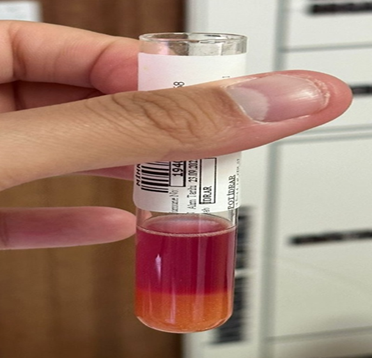
figure 1:
positive ehrlich aldehyde urine test
Methods
Diagnostic
evaluation included spot urine porphobilinogen testing with ehrlich's reagent,
confirming elevated levels suggestive of porphyria. Subsequent 24-hour urine
testing determined the specific subtype. Clinical correlation with cutaneous
manifestations and neurological history supported the diagnosis of porphyria.
Discussion
porphyrias are a group of inherited or acquired disorders characterized by a
defect in one of the eight enzymes involved in the heme biosynthetic pathway,
leading to the accumulation of porphyrins or their precursors. These
accumulations can result in a wide range of clinical manifestations, from acute
neurovisceral symptoms to chronic cutaneous lesions, depending on the specific
type of porphyria and the affected enzym1,2.
The complexity of porphyria's clinical presentation often leads to misdiagnosis
or delayed diagnosis, as seen in this patient, where the diagnosis was only
made after significant disease progression.
This
patient’s history of meningomyelocele spina bifida is a significant factor in
her overall clinical presentation. Spina bifida, particularly the
meningomyelocele subtype, is associated with a range of complications,
including neurological deficits, chronic urinary retention, and renal
dysfunction due to vesicourethral reflux3.
In this case, the patient’s renal failure began in her early twenties,
requiring escalating dialysis interventions. It is well-documented that chronic
renal failure can exacerbate the clinical manifestations of porphyria due to
the impaired clearance of porphyrins and their precursors4,5. This interplay between renal impairment and
porphyria likely contributed to the severity of the patient’s symptoms and the
difficulty in managing her condition.
The cutaneous
symptoms experienced by the patient, particularly the formation of bullous
lesions upon sun exposure (figures 2,3),
are characteristic of cutaneous porphyrias, such as porphyria cutanea tarda (pct)
or variegate porphyria (vp)6. These
lesions occur due to the accumulation of porphyrins in the skin, which become
photoactivated by ultraviolet light, leading to oxidative damage and blister
formation7. The chronicity and progression
of these lesions, resulting in permanent scarring, further underscore the
impact of delayed diagnosis and inadequate management. 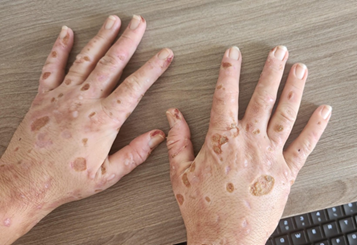
figure 2: lesions on hands,
parts exposed to sunlight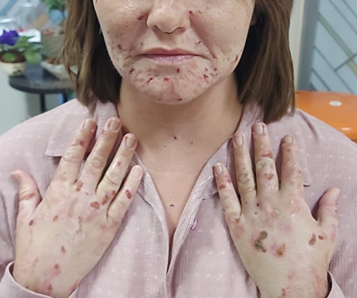
Figure 3:
lesions on sun exposed areas, both hands and face
Interestingly,
the patient’s family history, while not directly indicative of porphyria,
includes congenital adrenal hyperplasia in her mother and diabetes mellitus in
her father. Although these conditions are not directly linked to porphyria,
they may suggest a genetic predisposition to metabolic disorders, warranting
further investigation into potential familial links or genetic mutations
contributing to the patient’s condition8,9.
The absence of similar symptoms in immediate family members does not preclude a
hereditary basis, particularly in cases where porphyria may manifest with
varying severity or may remain asymptomatic in other family members.
The
diagnostic process for porphyria in this patient involved a spot urine
porphobilinogen test, which is a critical initial step in identifying acute
porphyrias10. Elevated levels of
porphobilinogen, along with clinical symptoms, strongly suggest a diagnosis of
porphyria. The subsequent 24-hour urine analysis is essential for measuring the
excretion of porphyrins and their precursors, helping to differentiate between the
various types of porphyria11. In this
case, the specific subtype is found to be vp, highlighting the need for genetic
testing to confirm the diagnosis and guide management.
Management
of porphyria, particularly in the context of concurrent renal failure, requires
a multidisciplinary approach. Dermatological care is necessary to manage the
cutaneous symptoms and prevent further skin damage, while nephrological support
is critical in managing the patient’s renal failure and ensuring the safe
administration of treatments12. Given
the complexity of this case, involving multiple systems and rare conditions, a
coordinated effort among specialists in neurology, nephrology, dermatology, and
genetics is essential to optimize patient outcomes.
Conclusion
In
conclusion, this case report highlights the challenges in diagnosing and
managing porphyria, particularly when it coexists with other congenital or
chronic conditions like spina bifida and renal failure. The delay in diagnosis
and the subsequent complications underscore the importance of early recognition
and intervention in patients with complex medical histories. Clinicians should
maintain a high index of suspicion for porphyria in patients with unexplained
dermatological and neurological symptoms, particularly when these symptoms are
accompanied by renal dysfunction or a history of congenital anomalies. Early
diagnosis and a multidisciplinary approach to care are essential to prevent the
progression of symptoms and improve the patient’s quality of life.
References
1. Anderson
ke, sassa s, bishop df, desnick rj. Disorders of heme biosynthesis: x-linked
sideroblastic anemia and the porphyrias. In: scriver cr, beaudet al, sly ws,
valle d, editors. The metabolic and molecular bases of inherited disease. 8th
ed. New york: mcgraw-hill 2001;2991-3062.
2. puy
h, gouya l, deybach jc. Porphyrias. Lancet 2010;375(9718):924-937.
3. bowman
rm, mclone dg, grant ja, tomita t, ito ja. Spina bifida outcome: a 25-year
prospective. Pediatr neurosurg 2001;34(3):114-120.
4. Singal
ak, anderson ke. Variegate porphyria. In: adam mp, ardinger hh, pagon ra,
wallace se, bean ljh, stephens k, et al., editors. Genereviews®.
Seattle (wa): university of washington, seattle. 1993-2021.
5. wahlin
s, floderus y, stal p, harper p. Erythropoietic protoporphyria in sweden:
demographic, clinical, biochemical and genetic characteristics. J intern med 2011;269(3):278-288.
6. elder
gh, hift rj, meissner pn. The acute porphyrias. Lancet 1997;349(9065):1613-1617.
7. phillips
jd, jackson lk, bunting m, et al. A porphomethene inhibitor of uroporphyrinogen
decarboxylase causes porphyria cutanea tarda. Proc natl acad sci usa
2001;98(17):9521-9526.
8. thadani
h, deacon a, peters tj. Diagnosis and management of porphyria. Bmj 2000;320(7250):1647-1651.
9. stein
pe, badminton mn, rees dc. Update review of the acute porphyrias. Br j haematol
2017;176(4):527-538.
10. marsden
jt, guppy s, stein p, et al. Audit of the use of hemin in the treatment of
acute porphyria in the uk. J clin pathol 2015;68(8):689-695.
11. gouya
l, puy h, lamoril j, et al. Inheritance of erythropoietic protoporphyria: a
common wild-type ferrochelatase allele is associated with clinical
manifestation. J clin invest 1999;103(7):987-992.
12. bonkovsky
hl, maddukuri vc, yazici c, et al. Acute porphyrias in the usa: features of 108
subjects from porphyria consortium. Am j med 2014;127(12):1233-1241.