Schistosomiasis-Associated Pulmonary Hypertension cured with Praziquantel Therapy
Abstract
This is a case report of a 52-year-old female who immigrated from Africa and
presented with impending right heart failure, characterized by shortness of
breath and leg swelling for the past two months. In the emergency room (ER), a
CT chest with a pulmonary embolism protocol was performed, which was negative
for clots but revealed findings of cirrhosis, splenomegaly and pulmonary artery
dilation. Further workup demonstrated iron deficiency anemia and severe
pulmonary hypertension, confirmed via right heart catheterization. Treatment
was initiated with diuretics and pulmonary vasodilators, eventually requiring
triple drug therapy that included intravenous epoprostenol. Given her travel
history, schistosomiasis was suspected and subsequently confirmed with antibody
testing, indicating chronic schistosomiasis with hepatic involvement. She was
treated with praziquantel but experienced a transient worsening of symptoms and
hypoxia due to hypersensitivity pneumonitis likely form dying parasites. The
patient responded well to a moderate dose of prednisone and was eventually
weaned off oxygen and epoprostenol. A three-month follow-up right heart
catheterization revealed near normalization of pulmonary pressures with
significantly improved functional capacity. Six months later, she was
successfully weaned off all pulmonary vasodilators.
Keywords: Impending right heart;
Epoprostenol; Schistosomiasis; Pulmonary vasodilators
Case Presentation
A
52-year-old non-alcoholic female with a medical history of hypertension and
type 2 diabetes presented with a two-month history of chronic pain, generalized
weakness, difficulty sleeping, chest discomfort, progressive exertional
shortness of breath and leg swelling. She reported a gradual decline in her
functional capacity over this period, eventually becoming unable to perform
activities of daily living. She also noted subjective evening fevers,
grogginess and at least two near-syncopal episodes prior to presentation. She
denied hemoptysis, hematemesis, weight loss, convulsions, wheezing or changes
in bowel or bladder habits.
The
patient had immigrated to the United States two months earlier and had not yet
established care with a primary physician. Upon arrival at the ER, her vital
signs were as follows: blood pressure (BP) 90/65 mmHg, heart rate (HR) 112/min,
respiratory rate (RR) 24/min and oxygen saturation (SpO2) 84%. Physical
examination revealed a distended abdomen, dilated superficial veins, jugular
venous distention (JVD) and pedal edema. Auscultation findings were
unremarkable. Oxygen supplementation was initiated at 4 L/min. Troponin levels
were negative for acute coronary syndrome, but an electrocardiogram (ECG)
showed multiple premature ventricular contractions (PVCs) with a right bundle
branch block (RBBB) pattern. A CT chest to rule out pulmonary embolism (PE) was
negative for clots but demonstrated cirrhotic liver changes, splenomegaly and
pulmonary artery dilation.
See (Figures
1,2 and 3).
Figure 1: Ct Chest axial view, mediastinal window demonstrating enlarged pulmonary
artery (yellow line) compared to aorta (red arrow) suggestive of pulmonary
hypertension

Figure 2: CT Chest Axial view, mediastinal window demonstrating dilated right
ventricle (red arrow) suggestive of pulmonary hypertension

Figure 3: Ct Chest at the level of liver, abdominal window
showing nodular configuration of Liver (red arrow) suggesting fibrosi
Laboratory results revealed a hemoglobin level of
7.6 g/dL, consistent with iron deficiency anemia; lactate level of 4.2 mmol/L;
serum creatinine of 1.5 mg/dL; and brain natriuretic peptide (BNP) of 400
pg/mL. She was admitted to the cardiac intensive care unit (ICU) with a
diagnosis of cardiogenic shock. Transthoracic echocardiography showed an
estimated pulmonary artery systolic pressure (PASP) of 81 mmHg and severely
reduced right ventricular systolic function, with a tricuspid annular plane
systolic excursion (TAPSE) of 1.1 cm. See (Figures
4 and 5).

Figure 4: Echocardiogram: Parasternal short axis view
demonstrating enlarged right ventricle (red arrow) compared to left ventricle
(green arrow) suggestive of pulmonary hypertension

Figure 5: Echocardiogram Top figure: Apical four chamber view revealing enlargement of right atrium (green arrow) and right ventricle (red arrow). Bottom figure: M mode of the Tricuspid annulus. TAPSE (tricuspid annular plane systolic excursion) was noted to be 1.1 cm denoting severe decrease in right ventricular function. Normal TAPSE is > 1.7 cm Iron supplementation and gentle diuretics were administered.
A ventilation-perfusion (VQ) scan showed no mismatch. See (Figure 6)

Figure 6: VQ Scan demonstrated mildly heterogenous perfusion and ventilatory radio-tracer distribution throughout both lungs (red arrows). No ventilation perfusion mismatch. Low probability of pulmonary embolism. Th over all pattern is suggestive of pulmonary hypertension
Right heart catheterization confirmed severe
pulmonary arterial hypertension (PAH), with findings including a mean pulmonary
artery pressure (mPAP) of 58 mmHg, pulmonary vascular resistance (PVR) of 16
Wood units (WU) and cardiac index (CI) of 1.95 L/min/m² with central venous
oxygen saturation of 35%. Vaso reactive testing showed a significant drop in
systemic blood pressure and cardiac output. Key catheterization data:
·
Baseline: RA 17 mmHg, PA 96/37/58 mmHg, PCW 4 mmHg, PVR 16
WU, CI 1.95 L/min/m²
· Drug study: RA 17 mmHg, PA 66/26/39 mmHg, PCW 4 mmHg, PVR 11 WU, CI 1.7 L/min/m
Treatment and Follow-Up: The patient was started on intravenous epoprostenol at 2 ng/kg/min, titrated to 10 ng/kg/min, with dose adjustments limited by side effects such as flushing. She was kept nil per os (NPO) pending an endoscopic evaluation. On day three, a gastroenterology consultation was obtained for cirrhosis, iron deficiency anemia and positive fecal occult blood tests.
Esophagogastroduodenoscopy (EGD) and colonoscopy
revealed grade 1 esophageal varices, portal hypertensive gastropathy and
multiple colonic ulcers, particularly in the sigmoid and rectum (Figures 7 and 8).
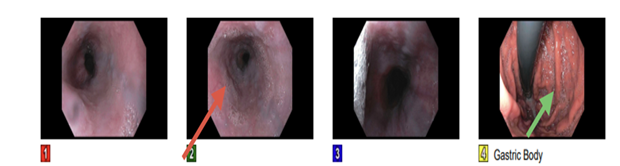
Figure 7: Upper GI endoscopy showed varices (red arrow) in the lower third of
esophagus and moderate portal hypertensive gastropathy in the gastric body
(green arrow). Duodenal biopsy revealed mildly increased intraepithelial
lymphocytes and intact villous architecture
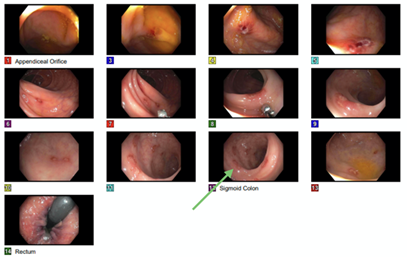
Figure 8: Colonoscopy showed multiple 2-6 mm ulcers in the entire colon with highest concentration in the sigmoid (green arrow) and rectum. Biopsy revealed active colitis with necro-inflammatory ulcer debris. There were no features of chronicity, granulomata or dysplasia. CMV immunostaining was negative. No viral inclusions noted. Serology was negative for Celiac disease and Inflammatory Bowel Disease (IBD)
Given her recent immigration from Africa, schistosomiasis was suspected. Testing confirmed chronic schistosomiasis with positive Schistosoma antibody and radiological evidence of cirrhosis, although stool ova and parasites were negative. Sildenafil was initiated on day four and uptitrated to 20 mg TID over 48 hours. On day six, macitentan 10 mg daily was added for triple therapy alongside epoprostenol and sildenafil, leading to significant improvements in functional status and hemodynamics. She was eventually weaned off oxygen. Praziquantel was administered in two doses, one week apart, as a part of anti-parasitic therapy. However, 24 hours after the first dose, the patient experienced worsening hypoxia and shortness of breath needing 4-6 l/min of Oxygen supplementation.
High-resolution CT chest revealed diffuse
ground-glass opacities with mosaic attenuation and pulmonary nodules,
suggestive of hypersensitivity pneumonitis. A short course of prednisone (40 mg
daily for seven days) resolved her hypoxia within 48 hours. See (Figure 9 )
Figure 9: Hi-resolution T Chest, lung window at the level of aortic arch demonstrating diffuse ground glass opacities (red arrow) with mosaic attenuation (green arrow) and nodularity (yellow arrow) after receiving anti-parasitic therapy. Responded very well to steroids
By day ten, epoprostenol was gradually weaned off
due to side effects of flushing and dizziness. The patient was maintained on
sildenafil and macitentan. Follow-up right heart catheterization showed
significant improvement, with a PA pressure of 29/48 mmHg, PVR of 6 WU and CI
of 2.48 L/min/m² and hence prostaglandin therapy was not -re-initiated as there
was good response to dual therapy associated with rapid improvement in her walk
distance. She was discharged on sildenafil 20 mg TID and macitentan 10 mg daily,
with a six-minute walk distance (6MWD) of 200 meters.
At the three-month follow-up, her 6MWD had increased to 500 meters and BNP levels had normalized to 45 pg/mL. Repeat echocardiography revealed normalization of pulmonary pressures. A repeat right heart catheterization showed further improvements with a PA pressure of 32/16/21 mmHg, a PCWP of 8 and a PVR of 2.3 WU. As she was slightly hypotensive and complained of mild orthostatic dizziness, macitentan was discontinued and Sildenafil was continued. Six months later, her functional capacity and BNP levels continued to improve and she remained asymptomatic. Sildenafil was gradually tapered off and she remained stable. The patient remained asymptomatic and continued to perform all activities without limitations. A repeat echocardiogram in 1 month after the 6 months follow up confirmed complete normalization of pulmonary pressures.
Discussion
Schistosomiasis, caused by parasitic trematodes, remains a
significant health concern in endemic regions such as Africa and Asia,
affecting approximately 200 million individuals globally1.
The chronic hepato-splenic form of the disease, a known risk factor for
pulmonary arterial hypertension (PAH), accounts for severe morbidity and
mortality2.
This case presents a striking example of schistosomiasis-associated pulmonary
arterial hypertension (Sch-PAH), a condition classified under World Health
Organization (WHO) Group 1 PAH (1.4.5)3. The
patient, a 52-year-old immigrant from Africa, presented with a constellation of
symptoms, including exertional dyspnea, lower extremity edema, syncope and iron
deficiency anemia. These features underscored the diagnostic challenge of
Sch-PAH, often overshadowed by clinical features of portal hypertension,
presumed liver cirrhosis and co-existing anemia.
The only clue to the diagnosis was the patient’s residence in an endemic region. Clinical findings included progressive exertional dyspnea, lower extremity edema, jugular venous distention (JVD) and a loud S2, which is highly suggestive of pulmonary hypertension and right ventricular dysfunction4. Initial evaluation revealed evidence of right ventricular dysfunction (TAPSE < 1.7 cm) with an elevated pulmonary artery systolic pressure of 81 mmHg on echocardiography, alongside severe pulmonary hypertension confirmed via right heart catheterization (mean pulmonary artery pressure of 58 mmHg and pulmonary vascular resistance of 16 Wood units).
It is crucial to distinguish between pre-capillary and post-capillary forms of pulmonary arterial hypertension, as the management differs significantly. Pulmonary vasodilators are indicated in the former but contraindicated in the latter. Right heart catheterization helps make this distinction. Pre-capillary pulmonary hypertension is defined by a mean pulmonary artery pressure (mPAP) > 20 mmHg at rest, pulmonary vascular resistance (PVR) > 2 Wood units and pulmonary capillary wedge pressure (PCWP) ≤ 15 mmHg. In contrast, post-capillary pulmonary hypertension, often associated with left heart disease, is characterized by a PCWP > 15 mmHg [3]. The patient’s immigration history and diagnostic imaging, which revealed cirrhosis and portal hypertension, raised suspicion for chronic schistosomiasis. Another important clue is the presence of eosinophilia in a patient with pulmonary arterial hypertension, which should prompt consideration of Sch-PAH2.
The diagnosis of Sch-PAH in this case was supported by positive Schistosoma antibody testing, even though stool ova and parasites were negative-a finding not uncommon in chronic disease due to intermittent egg shedding5. This case highlights the importance of integrating travel history and risk factor assessment into the evaluation of pulmonary hypertension, particularly in immigrant populations
The patient’s management demonstrated a dual therapeutic approach targeting both the underlying parasitic infection and pulmonary arterial hypertension. Anti-parasitic therapy with praziquantel was initiated at 60 mg/kg, administered in two doses one week apart, targeting the adult Schistosoma parasites. Notably, the patient developed hypersensitivity pneumonitis after the first dose, presenting as worsening hypoxia and ground-glass opacities with mosaic attenuation on computed tomography (CT). This inflammatory reaction, likely due to antigen release from dying parasites, was successfully managed with a short course of corticosteroids2.
Pulmonary arterial hypertension was treated with specific vasodilatory therapy. Initial management included intravenous epoprostenol, titrated according to tolerance, followed by the introduction of sildenafil and macitentan as part of a triple therapy protocol, given the high-risk assessment of her pulmonary hypertension. This combination led to marked clinical and hemodynamic improvement, with a reduction in PVR from 16 to 6 Wood units and normalization of pulmonary pressures over three months. The patient’s six-minute walk distance improved from 200 to 500 meters, reflecting a significant recovery in functional capacity.
Although many patients with Sch-PAH remain dependent on pulmonary vasodilator therapy indefinitely, this patient demonstrated significant hemodynamic improvement with anti-parasitic therapy6-9.
This suggests that some cases of Sch-PAH may be reversible.
Therefore, patients with positive Schistosoma antibodies and no prior
anti-parasitic treatment should be given the opportunity for therapy8,9.
The patient’s clinical course exemplifies the potential for substantial improvement in Sch-PAH with early and targeted interventions. Despite her initial presentation in cardiogenic shock, the combination of anti-parasitic therapy and pulmonary vasodilators resulted in sustained remission. Follow-up evaluations at six months revealed continued functional and hemodynamic improvement, normalization of biomarkers such as BNP (20 pg/mL) and cessation of vasodilator therapy without recurrence of symptoms.
Conclusion
This case
underscores the complex interplay between schistosomiasis, cirrhosis and
pulmonary hypertension. Early diagnosis and appropriate treatment with
praziquantel, combined with pulmonary hypertension-targeted therapy, led to
remarkable clinical improvement. Close monitoring and management of
treatment-related side effects, such as hypersensitivity pneumonitis, were
critical in ensuring a positive outcome. This case also illustrates the
potential for complete reversal of Sch-PAH with early treatment of schistosomiasis.
The use of the REVEAL score to guide risk stratification and management was
essential for tracking the patient’s progress and optimizing her treatment
regimen.
References
1. WHO. World Health
Organization 2025.
2. Pata R, Kosuru B, Kristeva J. Schistosomiasis-associated
pulmonary arterial hypertension (Sch-PAH): A comprehensive review of diagnosis
and management. Respiratory Medicine 2025;240:108026.
3. Humbert M, Kovacs G, Hoeper MM, et al. ESC/ERS Guidelines
for the diagnosis and treatment of pulmonary hypertension: Developed by the
task force for the diagnosis and treatment of pulmonary hypertension of the
European Society of Cardiology (ESC) and the European Respiratory Society (ERS).
European Heart J 2022;43:3618-3731.
4. Konstam
MA, Kiernan MS, Bernstein D, et al. American Heart Association Council on
Clinical Cardiology; Council on Cardiovascular Disease in the Young; and
Council on Cardiovascular Surgery and Anesthesia. Evaluation and Management of
Right-Sided Heart Failure: A Scientific Statement from the American Heart
Association. Circulation 2018;137(20):578-622.
5. Carbonell
C, Rodríguez-Alonso B, López-Bernús A, et al. Clinical Spectrum of
Schistosomiasis: An Update. J Clin Med 2021;10(23):5521.
6. apamatheakis DG, Mocumbi AOH, Kim NH and Mandel J.
Schistosomiasis-Associated Pulmonary Hypertension. Pulmonary Circulation 2014;4:596-611.
7. Sibomana
JP, Campeche A, Carvalho-Filho RJ, et al. Schistosomiasis Pulmonary Arterial
Hypertension. Front Immunol 2020;11:608883.
8. Crosby
A, Jones FM, Kolosionek E ,et al. Praziquantel reverses pulmonary hypertension
and vascular remodeling in murine schistosomiasis. Am J Respir Crit Care Med
2011;184(4):467-473.
9. Vale N, Gouveia MJ, Rinaldi G, et al. Praziquantel for Schistosomiasis: Single-Drug Metabolism
Revisited, Mode of Action and Resistance. Anti microb Agents Chemother 2017;61