Secondary Hemophagocytic Lymphohistiocytosis due to Anaplasmosis; When Infection meets Hyper-Inflammation: Consider Adjuvant Dexamethasone
Abstract
Hemophagocytic
Lymphohistiocytosis (HLH) is a rare, life-threatening hyper-inflammatory
condition characterized by excessive immune activation and multi-organ
dysfunction. HLH can be inherited (primary HLH) or acquired (Secondary HLH) due
to autoimmune disorders or infections. We report a case of a 72-year-old woman
who developed sepsis with HLH features secondary to Anaplasma phagocytophilum
infection. Despite appropriate antimicrobial therapy with doxycycline, she
experienced rapid clinical deterioration necessitating intensive care and vasopressor
support. Adjunctive treatment with a short course of intravenous dexamethasone
was associated with marked clinical improvement, resolution of vasopressor
dependence and recovery of organ function. This case highlights the importance
of early recognition of MAS in infectious contexts and supports consideration
of corticosteroid therapy alongside antimicrobials in selected patients.
Keywords: Anaplasma
phagocytophilum; Macrophage activation syndrome; Doxycycline; Dexamethasone; Multi-organ
dysfunction; Tick-borne infection, Hemophagocytic Lymphohistiocytosis
Introduction
Anaplasma phagocytophilum is an obligate intracellular
bacterium transmitted by ticks, causing human granulocytic anaplasmosis (HGA).
While most infections are self-limited or mild, severe cases may manifest with
systemic inflammation, cytopenia’s and organ dysfunction. Rarely, HGA can
trigger Hemophagocytic Lymphohistiocytosis (HLH) due to overwhelming immune
activation and cytokine storm resulting in clinical features suggestive of
sepsis, cytopenia’s and multi-organ dysfunction. HLH can be inherited (primary
HLH) or acquired (Secondary HLH) due to autoimmune disorders or infections1,2.
HLH should be suspected if the patient presents with
sepsis like syndrome along with cytopenia’s, liver dysfunction,
hyper-ferritinemia, coagulopathy and elevated inflammatory cytokines. Prompt
recognition and treatment are critical, as untreated HLH carries high mortality3,4. While antibiotics targeting the underlying infection
are essential, adjunctive immunomodulation with corticosteroids may be
lifesaving.
We present a case of 72-year-old woman with severe HLH
secondary to A. phagocytophilum infection successfully managed with doxycycline
and adjuvant dexamethasone.
Case Presentation
A 72-year-old previously
healthy woman presented with a 7-day history of progressive generalized
weakness and fever. On admission, she was febrile and appeared ill but
hemodynamically stable. Broad-spectrum antibiotics (vancomycin and meropenem)
were initiated pending diagnostic evaluation.
A detailed history obtained
from her spouse revealed a tick bite approximately 10 days prior, with no
prophylactic treatment. Peripheral blood smear demonstrated Howell-Jolly bodies
and intracellular inclusions consistent with Anaplasma species (Figure 1).
Doxycycline was started and broad-spectrum antibiotics were discontinued.
Serology was positive for Anaplasma phagocytophilum IgG antibody >1:2048 by
immunoblot.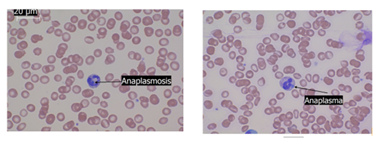
Figure 1: Peripheral blood smear
obtained on the patient revealed intracellular inclusions consistent with
Anaplasma. Also noted is Howell jolly bodies (nuclear remnants inside red blood
cell) and echinocytes (crenated red cells)
Within 12 hours of admission, the patient developed
profound hypotension requiring norepinephrine infusion and was transferred to
the intensive care unit. She experienced a lower gastrointestinal bleed and
worsening mental status, necessitating intubation. Computed tomography
angiography excluded active gastrointestinal bleeding or biliary obstruction.
Echocardiography showed hyper dynamic left ventricular function.
Laboratory evaluation revealed worsening liver enzymes
(AST 357 U/L, ALT 178 U/L, alkaline phosphatase 349 U/L, total bilirubin 5.9
mg/dL), acute kidney injury (serum creatinine 2.6 mg/dl), thrombocytopenia (60
×10^3/μL), prolonged partial thromboplastin time (54.6 sec) and coagulopathy
(fibrinogen 109 mg/dL, D-dimer 6940 ng/mL FEU). White blood cell count was 0.8
×10^3/μL. (Table 1) for laboratory values.
Table-1: Laboratory values of
our patient at the time of ICU admission
|
Test |
Patient
value |
Normal
range |
|
Lactate
Dehydrogenase |
868 U/L |
100-190 U/L |
|
Triglycerides |
750 mg/Dl |
<150 mg/dL |
|
Ferritin |
27,621 ng/mL |
13-150 ng/mL |
|
Interleukin-6
(IL-6) |
157 pg/mL |
<7 pg/mL |
|
Soluble IL-2
Receptor |
25,918.8 pg/mL |
223-710 pg/mL |
|
Aspartate
Transminase (AST) |
357 U/L |
10-40 U/L |
|
Alanine
Transminase (ALT) |
178 U/L |
7-56 U/L |
|
Alkaline
Phosphatase |
349 U/L |
44-147 U/L |
|
Total Bilirubin |
5.9 mg/dL |
0.1-1.2 mg/dL |
|
Serum creatinine |
2.6 mg/dl |
0.9 mg/dl |
|
White Blood Cell
Count |
0.8 ×103/μL |
4.0-10.5 ×103/μL |
|
Platelets |
60 ×103/μL |
150-450 ×103/μL |
|
Partial
Thromboplastin Time |
54.6 seconds |
25-35 seconds |
|
INR |
1 |
0.8-1.1 |
|
D-dimer |
6940 ng/mL FEU |
<500 ng/mL |
|
Fibrinogen |
109 mg/dL |
200-400 mg/dL |
Despite initiation of doxycycline, vasopressor
requirements increased over the subsequent 24 hours (norepinephrine up to 1.5
mcg/kg/min plus vasopressin 0.03U/min) and the patient remained oliguric with
worsening renal function (serum creatinine 2.6 mg/dl). Patient wes already
receiving hydrocortisone at 50 mg Q6hrs intravenously for refractory
hypotension. Further laboratory evaluation revealed elevated ferritin (27,621
ng/mL), triglycerides (750 mg/dL), lactate dehydrogenase (868 U/L). Due to
elevated triglycerides, ferritin, abnormal liver function tests and
cytopenia’s, secondary HLH was strongly suspected. Subsequently IL-6 came back
at 157 pg/mL and soluble IL-2 receptor was significantly elevated at 25,918.8
pg/mL strongly supporting a diagnosis of HLH or secondary macrophage activation
syndrome.
As patient had refractory hypotension and worsening
multi-organ dysfunction including acute kidney injury (AKI), on day 3, shared
decision was made with her husband and intravenous dexamethasone was initiated
at 10 mg/m² (23 mg), resulting in a rapid decrease in vasopressor requirements
within 4 hours. Norepinephrine was discontinued within 24 hours. Ferritin
decreased to 6415 ng/mL over the following days (Figure 2).
Figure 2: After administration of dexamethasone, ferritin levels decreased significantly with simultaneous improvement in vasopressor need
24 hours after intravenous dexamethasone, her
vasopressor requirements gradually increased over the next 12 hours needing
nor-epinephrine at 0.3 mcg/kg/min and hence a subsequent dose of dexamethasone
was given at 10mg/day. Following the administration of second dose of
dexamethasone, the nor-epinephrine could be weaned off. Dexamethasone was then
tapered over 3 days (10 mg followed by 8 mg), during which organ dysfunction
and laboratory indices improved significantly (Figure 3).
She never required renal replacement therapy during
the hospital course; however, required 48 hours of bumetanide infusion for
fluid management. Hyper-leukocytosis developed transiently but resolved within
48 hours. She was successfully extubated on day 8 and ultimately recovered
fully.
Figure 3: Effect of
dexamethasone on vasopressor need. Nor-epinephrine dose needed to maintain
target mean arterial pressure decreased gradually after administration of first
dose of dexamethasone and patient remained vasopressor free by 24 hours of
dexamethasone administration. However, in the next 12 hours, she required
progressively increasing doses of nor-epinephrine and hence received an
additional 10mg of dexamethasone with significant improvement in hemodynamics.
Subsequently, she was completely weaned off vasopressors and organ function
indices improved
Discussion
Anaplasma
phagocytophilum is an obligate intracellular bacterium transmitted by Ixodes
ticks, responsible for human granulocytic anaplasmosis (HGA). HGA typically
presents as a nonspecific febrile illness with symptoms such as fever, malaise,
myalgia, leukopenia, thrombocytopenia and mild hepatic enzyme elevation.
Although many cases are self-limiting or mild, severe disease with multi-organ
dysfunction can occur, particularly in elderly or immunocompromised patients1,2.
A rare but
life-threatening complication of HGA is secondary Hemophagocytic
Lymphohistiocytosis (HLH), also known as macrophage activation syndrome (MAS).
Secondary HLH/MAS is a hyper-inflammatory syndrome characterized by excessive
activation and proliferation of macrophages and cytotoxic T-cells, resulting in
a massive cytokine storm, multi-organ failure and high mortality if untreated3-5.
The pathogenesis of
HLH/MAS in infections such as Anaplasma involves immune dysregulation triggered
by persistent antigenic stimulation. Anaplasma infects neutrophils and evades
immune clearance by impairing phagocytic killing and modulating apoptosis pathways.
This leads to an exaggerated immune response with impaired cytotoxic function,
culminating in macrophage over activation and hemophagocytosis3,6,7.
Clinical and
laboratory features indicative of HLH/MAS include persistent high fever,
cytopenia's, hyper-ferritinemia, hyper-triglyceridemic, hypo-fibrinogenaemia,
elevated soluble IL-2 receptor (sCD25) and hemophagocytosis on bone marrow or
tissue biopsy. Although bone marrow biopsy may aid diagnosis, it is not always
definitive; clinical suspicion should guide early management3-5.
Our case illustrates
the potential for A. phagocytophilum infection to induce secondary HLH. The
initial presentation is like septic shock as seen in our case; however,
laboratory findings including bi-cytopenia, hyper-ferritinemia,
hyper-triglyceridemic and elevated soluble IL-2 receptors are suggestive of
secondary HLH or macrophage activation.
Although secondary
HLH/MAS triggered by A. phagocytophilum infection is rare, several case reports
describe this complication in elderly patients presenting with severe systemic
illness, including cytopenia's, liver dysfunction, coagulopathy and rapidly
progressing organ failure5,6. The diagnostic challenge arises because the
clinical picture mimics severe sepsis and delayed recognition of HLH/MAS can
adversely affect outcomes.
Whether secondary
HLH is considered a specific phenotype of sepsis or a separate entity mimicking
sepsis is a matter of perspective, but this condition needs to be identified.
As a typical sepsis, it should induce a robust Interleukin-6 response unlike secondary
HLH, as seen in our case where soluble IL-2 had a severe elevation compared to
IL-6. One should strongly suspect secondary HLH in any case of “sepsis” if
there are findings suggestive of bi-cytopenia, abnormal coagulation (low
fibrinogen and elevated d-dimer) and multi-organ dysfunction. Further
probability can be gauzed by ferritin and triglyceride levels as abnormal
coagulation and multi-organ dysfunction can be signs of sepsis or disseminated
intravascular coagulation (DIC). Of note, DIC can be a feature of HLH. If
needed IL-2R (also referred to as soluble CD25) and IL-6 can be obtained that
will help to identify this sub-phenotype of secondary HLH presenting as sepsis.
(Figure 4) for proposed flowchart for the diagnosis of secondary HLH.
Figure-4: Proposed flow chart for the
diagnosis of secondary HLH in suspected septic shock patients. See text for
further description. The cut-offs used in this algorithm are higher than the
traditional values used for the diagnosis of HLH, as sepsis by itself can
increase the levels of d-dimers, ferritin, LDH. These cut-off values represent
author’s personal opinion. IL-6 will be elevated from baseline and relatively
low compared to IL-2R
Abbreviations
PBS: peripheral blood smear,
SOFA: Sequential organ failure assessment score, IL: Interleukin, LDH: Lactate
dehydrogenase.
Our case demonstrates this severe
hyper-inflammatory phenotype. Despite appropriate antimicrobial therapy, the
patient’s condition deteriorated with increasing vasopressor requirements,
worsening cytopenia’s, markedly elevated ferritin and other biochemical markers
consistent with HLH.
While doxycycline is the
treatment of choice for Anaplasma, adjunctive immunosuppressive therapy,
particularly corticosteroids, has been employed successfully in severe cases to
dampen the hyper-inflammatory response6,7.
Standard treatment of HGA
involves doxycycline; however, in cases complicated by HLH, antimicrobials
alone may not suffice to arrest the cytokine storm and hyper-inflammation. Our
patient demonstrated a dramatic clinical response to corticosteroids, consistent
with other reports emphasizing immunosuppressive therapy in secondary HLH/MAS
triggered by infections. Of note there was significant temporal variation in
the vasopressor need with the administration of dexamethasone. The rapid
clinical improvement following initiation of dexamethasone supports its role in
interrupting the cytokine storm and immune-mediated tissue damage. In our case,
administration of dexamethasone resulted in significant clinical improvement,
including rapid reduction of vasopressor needs and recovery of organ function.
Corticosteroids down regulate the
excessive macrophage and T-cell activation driving HLH3. There is always a concern of
worsening infection with corticosteroids and hence only a short and tapering
course of steroid was employed in our case. The short course of dexamethasone
used in this case was well tolerated and temporally correlated with clinical
stabilization and improvement in vasopressor requirements.
Corticosteroids are typically
first-line agents due to their anti-inflammatory effects and safety profile for
short-term use. Dexamethasone is the preferred anti-inflammatory agent in this
condition as hydrocortisone due to its weak anti-inflammatory action alone may
not be sufficient. Other therapies, such as etoposide or cytokine inhibitors
(e.g., anakinra), are reserved for refractory cases or primary HLH3,4.
The rarity of Anaplasma-induced
HLH/MAS means current management is largely based on case reports and expert
opinion. Large prospective studies are needed to define optimal
immunosuppressive regimens, corticosteroid dosing and duration and time of
initiation. Identification of reliable biomarkers to predict progression to
hyper-inflammatory states could facilitate earlier diagnosis and improve
patient outcomes. We recommend obtaining ferritin in patients with multi-organ
dysfunction and new onset bi-cytopenia. If ferritin is > 1000 ng/ml, HLH
should at least be in the differential. Further assessments can be done by
fasting triglyceride levels, Il-2 and Il-6 levels.
Conclusion
Physicians should maintain a high
index of suspicion for HLH in patients with tick-borne infections who
deteriorate despite appropriate antimicrobial therapy, especially in the
presence of cytopenia’s, coagulopathy and hyper-ferritinemia. Early diagnosis
and initiation of adjunctive corticosteroids (for example dexamethasone)
alongside doxycycline can be lifesaving. Further research is warranted to
define optimal immunomodulatory strategies in infection-associated MAS.
References
1. Bakken JS, Dumler S. Human
granulocytic anaplasmosis. Infect Dis Clin North Am 2008;22(3):433-448.