The Dilemma of Management of 5-Alpha Reductase Enzyme Two Deficiency: A Single Clinic Experience
Abstract
Background: 5-alpha-reductase type 2 enzyme deficiency (5ard), is a rare autosomal recessive disorder of sex devolvement. The lack of the enzyme that converts testosterone into dihydrotestosterone, results in undervirilization of the external genitalia, ranging from phenotypic female to variable penile hypospadias.
Material and methods: this is a retrospective study which describes a series of twelve patients with 5-α- reductase deficiency. Data extracted from the medical records, included: history, clinical presentation, the appropriate, radiological and hormonal, investigations and management.
Results: During the period under review, a total of 69 patients with 46 xy dsd were evaluated. Twelve (20.3%) patients were proved to have 5α reductase deficiency. Their ages ranged from birth to 12 years. All patients presented with atypical appearance of external genitalia, ranging from clitoromegaly, isolated microphallus with penile hypospadias and undescended testes, to a completely normal looking female genitalia. Four (33.3%) patients had severe undervirilization, therefore, assigned female sex at birth. The other eight (66.6%) were given male sex. Unfortunately no genetic studies were done due to unavailability.
Conclusion: Disorders in male sexual differentiation result in an incompletely masculinized individual with a 46 xy karyotype and testes. Every effort should be made to accurately diagnose and manage this rare disorder, 5α reductase deficiency and its variable presentation and course. Given the complexity and nature of the disorder, it's extremely important to utilize coordinated multidisciplinary team of specialists. An interdisciplinary support is needed throughout childhood and adolescence.
Keywords: 5-alpha-reductase deficiency, disorder of sex development (dsd), 46xy karyotype, sex-reassignment.
Introduction
Disorders
in male sexual differentiation result in an under masculinization
(undervirilization) of an individual with a 46 xy karyotype and testes (figure
1). Various genetic mutations in the enzyme 5 α reductase which required to
metabolize testosterone (t) to dihydrotestosterone (dht). It is a rare disease
inherited as an autosomal recessive disorder with world-wide distribution. It
is 5-α- reductase deficiency that results in variable degrees of
undervirilization, ranging from typical female external genitalia to penile
hypospadias or isolated micropenis. The uterus and fallopian tube are absent,
due to normal production of mullerian inhibiting factor (mif). Testes are
intact and usually found in the inguinal canal or scrotum and wolffian ducts
differentiation is normal. With puberty, the affected males have increased
muscle mass, deepening of the voice and there is no gynecomastia. There is
substantial growth of the phallus, with rugation and hyperpigmentation of the
scrotum1-6. Patients could be
mistakenly diagnosed as having partial or complete androgen insensitivity
syndrome (ais) which can produce almost identical phenotypes. Every effort
should be made to accurately diagnose a newborn with ambiguous genitalia,
associated with 5-α-reductase-2-enzyme deficiency. A coordinated
multidisciplinary team of specialists should be utilized. The team consists of
pediatric endocrinologist, pediatric radiologist, geneticist, pediatric
surgeon, urologist, plastic surgeon, child psychologist or psychiatrist and
other specialties such as a nurse and gynecologist to be consulted whenever
needed7.
Figure 1: a medical photograph of a newborn infant with normal appearing external female genitalia and clitoromegaly. He had 46 xy karyotype and diagnosed with 5-α- reductase type 2 enzyme deficiency.
This article focuses on the 5-α-reductase-2-deficiency syndrome diagnosis and management and its impact on sex reassignment as seen over 30 years period at the king khalid medical city (kkmc), king saud university (ksu), riyadh, saudi arabia. The kkmc is the main teaching institute of ksu and considered as one of the main referral hospitals in riyadh, central province of saudi arabia. It provides primary, secondary and tertiary health care services for the local population and receives patients referrals from all over the country.
Material and methods
Patients with 46 xy dsd, due to 5-α-reductase 2 deficiency who were evaluated and managed at the kkmc, ksu, riyadh, saudi arabia over 30 years period were included in this retrospective study. The diagnosis was established based on hormonal investigations, post-human chorionic gonadotrophin (hcg). Dihydrotestosterone (dht), to testosterone (t) ratio was high in all patients8-10.
The medical records of patients were reviewed. Data included history, clinical presentation, the appropriate, radiological and hormonal, investigations and the management including the sex assignment. The ethical approval for this study was obtained from the institutional review board (irp), at king khalid university hospital.
Results
During the period under review, a total of 69 patients with 46 xy dsd were evaluated. Twelve (20.3%) patients, were diagnosed to have 5-α-reductase deficiency, based on hormonal investigations, the (dht/t) ratio, post-hcg stimulation, was high in all patients. Their ages ranged from birth to 12 years. The diagnostic imagings (ultrasound and/or magnetic resonance mr) were utilized to elucidate the internal organs (figures 2 and 3). None of the patients were found to have internal female structures. However all were found to have testes in variable positions (abdomen, inguinal canal and scrotum).
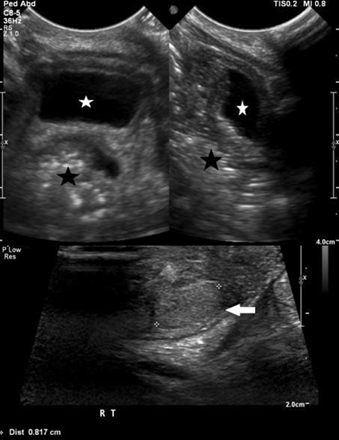
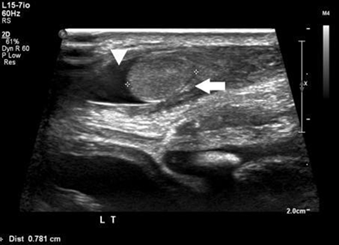
Figure 2: ultrasound pelvis (a) and scrotum (b) and (c) demonstrate absence of uterus and ovaries with presence of both testes in the scrotum (white arrows) with mild hydrocele on left side (white star). The urinary bladder (white star) and rectum (black star).
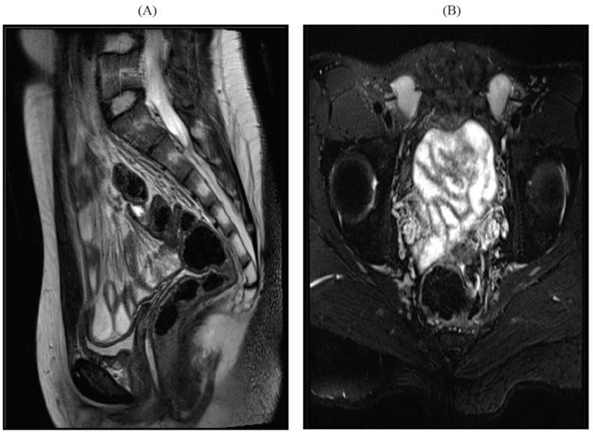
Table 1: clinical characteristics in twelve patients with 5 a-reductase deficiency
|
Patients |
Age at initial
diagnosis |
Given sex |
Clinical features |
Sex
of rearing and age |
Family history |
Remarks |
|
1 |
Birth |
Male |
Ambiguous genitalia, micropenis with chordee and bilateral undescended testicles |
Male - 4 days |
-ve |
|
|
2 |
Birth |
Male |
Hypospadias with
chordee |
Male - 4 days |
-ve |
|
|
3 |
Birth |
Male |
Unilateral undescended testicles hypospadias |
Male - 4 days |
-ve |
|
|
4 |
12 years |
Female |
Normal appearing
female genitalia with clitoromegaly |
Male - 12 years |
+ve |
|
|
5 |
6 months |
Male |
Hypospadia with bilateral undescended testicles |
Male - at birth |
-ve |
|
|
6 |
3 months |
Male |
Hypospadia with
bilateral undescended testicles |
Male - at birth |
-ve |
|
|
7 |
8 years |
Female |
Normal appearing
female genitalia |
Male - 8 years |
+ve |
|
|
8 |
4 years |
Female |
Urogenital
sinus with bifid, empty scrotum |
Male
- 4 years |
-ve |
|
|
9 |
1 year |
Male |
Micropenis with bilateral undescended testicle |
Male - at birth |
-ve |
|
|
10 |
Birth |
Female |
Normal appearing female genitalia with
clitoromegaly |
Male - 1 month |
-ve |
|
|
11 |
6 weeks |
Male |
Hypospadia with bilateral undescended testicle |
Male – 2 weeks |
-ve |
|
|
12 |
Birth |
Male |
Micropenis with bilateral undescended testicle |
Male – 2 weeks |
-ve |
|
All patients revealed no female internal structures with testis in variable positions (abdomen, inguinal canal and scrotum) by ultrasound or mri, +ve (positive), -ve (negative).
Four (33.3%) patients (two were siblings) had severe undervirilization and therefore wrongly assigned female sex. All were reassigned male sex at a later age. Unfortunately no genetic studies were performed, due to unavailability.
Discussion
Disorders
in male sexual differentiation are presently classified into three major
categories: gonadal development defects, such as gonadal dysgenesis, gonadal
biosynthesis and metabolism defects, such as 5-α-reductase deficiency and
gonadal disorders related to androgen insensitivity syndrome (ais).
5-α-reductase type 2 enzyme deficiency have variable phenotypes, ranging from
typical female external genitalia at birth, to more or less incomplete
virilization of the external genitalia, as in this series. Four patients had
severe abnormalities of their external genitalia, (undervirilization) and were
wrongly assigned a female sex, (table 1, patients 4,7,8 and 10).
Establishing the diagnosis of 5-α-reductase deficiency is often suggested by an elevated plasma testosterone (t), to dihydrotestosterone (dht) ratio following human chorionic gonadotropin hormone (hcg) stimulation8-10. Genetic studies can confirm the diagnosis11-16. Imaging diagnostic studies with ultrasonography (us) and/or magnetic resonance (mr) will help in elucidating the internal structures. In all patients no female internal organs were demonstrated, however, all were found to have testes in variable positions (abdomen, inguinal canal and scrotum)17-24.
In a community, like saudi arabia with increased prevalence of consanguineous marriage and multiple siblings, it is not unusual to see such numbers of patients25-27. Therefore, 5-α-reductase deficiency should be considered in the diagnosis of individuals with 46 xy dsd.
Theoretically, masculinization of the brain occurs under the influence of testosterone during the prenatal and neonatal periods. The development of a male gender identity occurred. Therefore, a male gender should be assigned at birth or reassigned later. Furthermore virilization at puberty, a hallmark feature of the disorder, will occur28-32. Many of those individuals will be fertile and the possibility of fatherhood are the main indicators for male sex assignment1,3,6,33-37. Gender identity and gender role may be a dynamic process that is not complete until well after adolescence. Four patients in this series, were mistakenly assigned female sex being presented in the neonatal period with near normal female external genitalia. Two of them were siblings where the older brother showed a clinical evidence of virilization at puberty. All were assigned male sex (table 1).
Every effort should be made to accurately diagnose and manage this rare disease. A well-coordinated team of specialists, consists of pediatric endocrinologist, pediatric radiologist, geneticist, pediatric surgeon, urologist, plastic surgeon, child psychologist or psychiatrist and other specialist such as a nurse and gynecologist to be consulted when ever needed. An interdisciplinary physiological support is needed throughout childhood and adolescence where all the available information should be progressively disclosed38-45.
Acknowledgement
The authors would like to thank mr. Ibrahim na aljurayyan, medical student, for his kind assistance in
preparing and typing this manuscript.
Conflict
of interest
The authors have no
conflicts of interest to declare.
Funding
None.
References
1. imperato-mcginley
j, guerrero l, gautier t, peterson re. 5 alpha-reductase deficiency,
curr ther. Endocrinol metab 1974;186(4170):1213-1215.
2. Aljurayyan n, aljurayyan a,
alissa s, mohamed s, alotaiabi h, babiker a. 5-alpha-reductase deficiency syndrome: an experience from a referral university hospital in saudi arabia pnco
2015;3(1):113-117.
10. Styne dm, the testes,
disorders of sexual differentiation and pberty.
In kaplan sa. Ed clinical pediatric endocrinology philadelphia, wb saunders co 1990:367-405.
17. Wherrett dk. Approach to
the infant with a suspected disorder
of sex development. Pediatr clin north am. 2015;62(4):983- 9.89.
18. al jurayyan na; imaging of disorders of sex development. Ann saudi med
2013;33(4):363-367.
42. cheon ck.
Practical approach to steroid 5-a-reductase type 2 deficiency. Eur j pediatr 2011;170(1):1-8.
43. mendonca bb, inacio m, costa em, et al. Male pesudohemaphrodisitism due to steroid 5alpha- reductase 2 deficiency. Diagnosis, psychological
evaluation and management.
Medicine (baltimore)
1996;75(2):64-76.
44. sharma s, gupta
dk. Male genitoplasty for intersex disorders.
Adv urol. 2008;4,589-605.