From Complexity to Simplicity: AI's Route Optimization in Supply Chain Management
S. Elouahab*, L. Laghsene, S. Bensmimou, M. Lahjaouj, M. Loudghiri, W. Bijou, Y. Oukessou, S. Rouadi, R. Abada, M. Roubal and M.
Mahtar
Department of Otorhinolaryngology, Head and Neck Surgery (ENT), 20 August Hospital, CHU Ibn Rochd, Casablanca, Morocco.
Citation: Elouaha S, Laghsene L, Bensmimou M, etal. Parathyroid Carcinoma Presenting with Severe Primary Hyperparathyroidism and
Osteolytic Skeletal Lesions: A Case Report. Medi
Clin Case Rep J 2025;3(4):1542-1545. DOI: doi.org/10.51219/MCCRJ/
Elouahab-Sara/425
Received: 18 December, 2025; Accepted: 24 December, 2025; Published: 26 December, 2025
*Corresponding
author: Elouahab Sara, Department of Otorhinolaryngology, Head and Neck
Surgery (ENT), 20 August Hospital, CHU Ibn Rochd, Casablanca, Morocco
Copyright:
©
2025 Elouaha S, et al., This is an open-access article distributed under the
terms of the Creative Commons Attribution
License, which permits unrestricted use, distribution, and reproduction in any
medium, provided the original author and source are credited.
A B S T R A C T
Parathyroid carcinoma is an exceptionally rare malignancy typically presenting with severe primary hyperparathyroidism. Diagnosis is often difficult and relies mainly on histopathological evaluation. Surgery remains the cornerstone of treatment. We report the case of a 52-year-old man with chronic right hip pain and anxiety disorder, who presented with progressive general deterioration. Clinical examination revealed a firm, painless, anterior left-lateral basocervical mass measuring approximately 6 cm. Laboratory investigations showed marked hypercalcemia (144 mg/L) and severe hyperparathyroidism with a parathyroid hormone (PTH) level of 1,737 pg/mL (39 times normal). Imaging studies demonstrated a large heterogeneous mass adjacent to the lower pole of the left thyroid lobe, associated with multiple osteolytic skeletal lesions.
A giant parathyroid adenoma was suspected and surgical excision was performed after preoperative correction of hypercalcemia. The mass extended into the retrosternal space without invasion of adjacent structures. Intraoperative histology suggested adenoma or hyperplasia. Postoperatively, calcium and PTH levels normalized. However, definitive histopathological analysis confirmed parathyroid carcinoma, showing capsular invasion and vascular emboli.
At the six-month follow-up, the patient showed
significant improvement in bone pain,
with no evidence
of recurrence and normal
calcium levels. This case highlights the diagnostic difficulty of parathyroid
carcinoma and underscores the need for long- term surveillance due to the risks of recurrence and metastasis.
Keywords: Parathyroid; Neoplasms; Carcinoma; Hyperparathyroidism; Primary; Hypercalcemia; Parathyroidectomy
Introduction
Parathyroid
carcinoma (PC) is an exceptionally rare endocrine malignancy, accounting for fewer than 1% of all cases of primary hyperparathyroidism
(pHPT)1. The condition was first documented in 1904 by the Swiss surgeon de Quervain2 in a
patient presenting with a non-functioning parathyroid lesion. Since this
initial description, the global literature has progressively expanded our
understanding of this uncommon neoplasm.
In this article, we describe a case of parathyroid carcinoma in a patient with persistent pHPT, reported in accordance with the SCARE criteria3 and provide a concise review of the relevant literature.
Presentation of Case
A 52-year-old man with a history of right hip pain and anxiety disorder for
the past three years presented with progressive deterioration of his general
condition. Physical examination revealed a firm, painless, anterior
left-lateral Baso cervical
mass, mobile on swallowing, measuring approximately 6 cm in its greatest dimension, with a non-palpable inferior border (Figure
1). No cervical lymphadenopathy was detected and vocal cord mobility was
preserved.
Laboratory
investigations demonstrated marked hypercalcemia (144 mg/L) and severe hyperparathyroidism,
with an extremely elevated parathyroid hormone (PTH) level of 1,737 pg/Ml-approximately 39 times the
upper limit of normal. Standard skeletal radiographs revealed diffuse bone
demineralization, predominantly affecting the iliac bones, femoral necks and
the right femoral diaphysis.
Cervical
ultrasonography identified a heterogeneous, hypoechoic and hypervascular mass
located adjacent to the lower pole of the left thyroid lobe, measuring 62 × 53 × 42 mm.
Cervico-thoracic computed tomography (CT) confirmed a well-
defined, solid-cystic oval lesion beneath the left thyroid lobe, with smooth
margins, measuring 54 × 45 mm and extending 63 mm in height. The mass showed close anatomical
relations with the left common carotid artery, situated anterior to it.
Additionally, multiple costal and vertebral osteolytic lesions were noted (Figure 2). Abdominopelvic CT revealed
multiple osteolytic lesions involving the spine and pelvis.
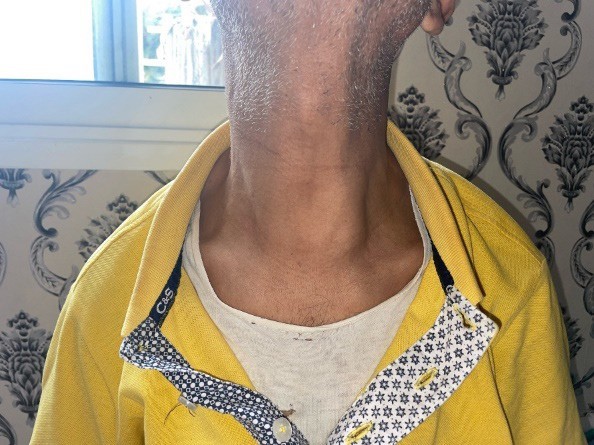
Figure 1: Physical examination revealed anterior left-lateral Baso cervical mass, mobile on swallowing, measuring approximately 6 cm in its greatest dimension, with a non-palpable inferior border.

Figure 2: Axial
abdominopelvic CT scan showing osteolytic bone
lesions involving the spine and the pelvis
Based on
these findings, a parathyroid adenoma was suspected and surgical excision was
planned. Preoperative management included intravenous rehydration with isotonic
saline and administration of bisphosphonates to correct the hypercalcemia.
Intraoperatively, the mass was found to be plunging into the retrosternal space, with no evidence of infiltration or continuity with the lower pole of the left thyroid lobe, which appeared macroscopically normal. A left inferior parathyroidectomy was performed and the excised specimen was sent for intraoperative histopathological examination, which suggested either an adenoma or parathyroid hyperplasia without features of malignancy.
The immediate
postoperative course was uneventful. Biochemical assays on postoperative day 1
demonstrated normalization of serum calcium (103 mmol/L) and PTH levels (44
pg/mL).
Definitive histopathological analysis established the diagnosis of parathyroid carcinoma, characterized by an encapsulated malignant proliferation composed of parathyroid cells arranged in diffuse and nodular patterns, separated by fibrous septa. The tumor cells were monomorphic, with abundant cytoplasm and mildly atypical round nuclei. Mitotic activity was moderate (three mitoses per ten high-power fields). Areas of capsular invasion and vascular tumor emboli were also identified.
At the six-month follow-up, the patient showed significant improvement in bone pain, with no clinical or ultrasonographic evidence of local recurrence. Laboratory findings confirmed normalization of serum calcium levels.
Discussion
Parathyroid carcinoma
(PC) is an extremely rare endocrine malignancy, representing
less than 0.005% of all cancers and accounting for approximately 0.5-4% of
primary hyperparathyroidism cases, with significant geographical variation
reaching up to 5% in Japan4,5. In
Western countries, PC usually accounts for less than 1% of pHPT cases6. Its incidence is estimated at 4-6 cases
per 10 million inhabitants per year and a large American series reported 286
cases over 10 years7,8. Because of its rarity and lack of
specific clinical and biological
signs, PC is frequently misdiagnosed as benign primary hyperparathyroidism and
is often diagnosed only postoperatively4,7,9,1.
The etiology
of PC remains poorly understood, although several environmental and genetic
factors have been implicated10,7,9.
Neck irradiation, particularly at a young age, increases the risk of
parathyroid neoplasia5,6.
Chromosomal abnormalities-including 1p, 4q, 13q losses and 1q, 9q, 16p, Xq
gains-have been reported11 and cyclin D1 overexpression is found in most tumors12. Parathyroid carcinoma also shows a strong
association with hyperparathyroidism–jaw tumor syndrome13. Additional genetic
abnormalities, such as RB, p53, BRCA2 and PRAD1 mutations, have been
described13.
Most PCs are functioning tumors causing severe hypercalcemia, presenting with fatigue, weakness, weight loss, anorexia, psychiatric symptoms, gastrointestinal complaints, nephrolithiasis and bone lesions including brown tumors10,14,15. Renal and skeletal involvement is common at presentation16.
Dysphonia and
dysphagia, resulting from recurrent laryngeal nerve invasion, are highly suggestive of malignancy10. Marked hypercalcemia-often above 3.5
mmol/L-is frequently observed11,17.
Non-functioning carcinomas are extremely rare and typically present with
advanced local disease7,15,18.
Imaging plays
a key role in the evaluation of PC. Cervical ultrasound may show lobulated
hypoechoic lesions with irregular margins, intra-lesional calcifications or
infiltration of adjacent tissues-features suggestive of malignancy10,15. Negative
predictive features include a thick capsule, ovoid shape or absence of
intratumoral vascularity10.
Ultrasound sensitivity ranges from 50% to 90%15.
Tc-99m sestamibi scintigraphy is useful for localization but cannot distinguish
adenoma from carcinoma; however, it may detect lymph-node or distant metastases15,19. CT and MRI offer better visualization
of soft tissue invasion and nodal involvement14,20. FDG-PET
may show uptake in brown
tumors, which can mimic metastasis21.
Fine- needle aspiration cytology is not recommended due to false negatives and
risk of capsular rupture22,23.
Intraoperatively,
PC typically appears as a firm, lobulated mass with a dense gray-white fibrous
capsule adherent to surrounding tissues, making
dissection difficult17,24. Tumors are usually large (>3 cm) and may
involve adjacent structures25.
Histopathological diagnosis is difficult. Classic criteria include trabecular
architecture, fibrous bands, mitotic activity and capsular or vascular invasion7,1,26, but these features are not specific
and may also occur in benign lesions27.
Surgery is
the mainstay treatment for PC. Recommended management includes en bloc
resection of the tumor with ipsilateral thyroid lobectomy and excision of
involved lymph nodes14,15,28. Complete
excision provides the best chance
of cure, while incomplete resection is associated with recurrence7,14,15,24. Avoiding capsular
rupture is essential to prevent tumor seeding10. Lateral lymph-node dissection is
recommended only when nodal metastases are present28,29.
Although PC is traditionally considered radioresistant, radiotherapy may
improve local control in selected cases17,20.
Chemotherapy has not shown proven benefit7,10.
Recurrence is
frequent, occurring in 25% to 60% of cases within the first 2-5 years7,30. Late recurrences, sometimes beyond
20 years, have been reported, requiring prolonged follow-up7. Recurrence often presents with rising
serum calcium and PTH levels and may involve local, regional or distant
metastases31. Follow-up includes
physical examination and serial monitoring of calcium and PTH13. Management of hypercalcemia may require
loop diuretics, dialysis or bisphosphonates10,14.