Anisotropic Growth of Doublet and Triplet Silica Colloids in a Butanol–PVP System
Abstract
The controlled fabrication of anisotropic silica colloids such as doublets and triplets remains a significant challenge in colloidal materials chemistry due to the tendency of silica to form isotropic spherical particles under classical conditions. Herein, we report a solvent-modulated and polymer-assisted strategy for synthesizing discrete doublet and triplet silica particles using a modified Wilhelm Stöber-type reaction conducted in n-butanol in the presence of PVP. Replacement of ethanol with n-butanol reduces hydrolysis kinetics of TEOS, lowers dielectric screening and increases particle-particle interaction time. Concurrently, partial surface adsorption of PVP generates anisotropic shielding, enabling controlled neck formation upon collision. By tuning TEOS feed rate, ammonia concentration, PVP molecular weight and particle density, discrete doublet and triplet colloids were achieved with minimal higher-order aggregation. Further anisotropic morphology of organosilica from TPM was observed and studied briefly. Electron microscopy confirms silica neck growth at contact interfaces, supporting a condensation-driven fusion mechanism. The presented method offers a scalable, solution-based route toward colloidal molecules suitable for directional self-assembly, photonic structures and hierarchical material design.
Introduction
Monodisperse
silica nanoparticles synthesized via the classical Wilhelm Stöber process1 have been extensively employed in
catalysis, photonics, sensing and surface science. In its conventional form,
base-catalyzed hydrolysis and condensation of Tetraethyl orthosilicate (TEOS)
in ethanol-ammonia media yields highly uniform spherical particles. The
isotropic nature of nucleation and growth, combined with strong electrostatic
stabilization in polar solvents, typically prevents controlled anisotropic
assembly.
However, colloidal
clusters such as doublets (two fused spheres) and triplets (three fused
spheres) are of increasing interest because they function as “colloidal
molecules2” with directional
bonding and tunable valency. Such structures are valuable in photonic bandgap
materials, programmable self-assembly and anisotropic coating technologies. Achieving
discrete fused clusters rather than uncontrolled aggregation requires precise
control over of hydrolysis and condensation kinetics, interparticle
electrostatic repulsion, collision frequency and surface stabilization Solvent
polarity plays a fundamental role in modulating reaction kinetics and particle
stabilization. Ethanol promotes rapid TEOS hydrolysis and strong charge
stabilization, limiting particle fusion. In contrast, n-butanol possesses lower
polarity and higher viscosity, which slows hydrolysis and reduces electrostatic
repulsion.
Polymeric
additives such as Polyvinylpyrrolidone (PVP) are known to adsorb onto silica
surfaces via hydrogen bonding between its carbonyl groups and surface silanol (Si-OH)
groups. Depending on coverage and molecular weight, PVP can act as steric
stabilizer, bridging agent and as anisotropic surface modifier.
In this work, we
demonstrate that combining solvent polarity reduction with controlled PVP
adsorption enables directional fusion and formation of doublet and triplet
silica colloids in a scalable batch process.
Experimental
Section
Materials
Tetraethyl
orthosilicate (TEOS),3-(Trimethoxysilyl) propyl methacrylate (TPM) Polyvinylpyrrolidone
(Mw 40,000), Sorbitan Monooleate (Span 80), Cetyltrimethylammonium bromide
(CTAB),
Potassium persulfate (KPS),
n-Butanol, Ammonium hydroxide solution (25-28 wt%) from Sigma Aldrich and
Deionized water (DI). All reagents were used as received.
Synthesis of silica seed particles
A
typical reaction mixture contained with n-butanol (solvent phase) and ammonium
hydroxide as catalyst. 0.1wt% TEOS was added dropwise under moderate stirring
at room temperature. Once the solution turns turbid PVP was introduced at
concentrations between 0.05 wt%. A secondary controlled feed of TEOS at same
concentration of 0.1wt% was added slowly. Reaction goes on for overnight Particles
were purified by centrifugation and redispersed in fresh n-butanol. SEM and TEM
characterisation was conducted to analyse the morphology of particles.
Synthesis of
multifunctional silica colloidal molecules
A precursor
emulsion was first prepared by mixing 1 mL of TPM with 0.1 mL of DI in the
presence of 0.03 wt% Span 80 under vigorous stirring to obtain a stable water-in-oil
type emulsion. This emulsion was subsequently dispersed into 10 mL of n-butanol
containing 0.05 wt% CTAB to enhance interfacial stabilization. The resulting
precursor mixture was then introduced into an equi-volume solvent system (100
mL total) composed of n-butanol and DI, containing 1 wt% ammonium hydroxide
solution (28% w/v in water), under continuous stirring at 72 °C. The basic
environment facilitated the hydrolysis and condensation of TPM. After 15
minutes of reaction, 2 mL of an aqueous initiator solution of KPS (0.01% w/v)
was added to initiate free-radical polymerization of the methacrylate groups.
The reaction was allowed to proceed for 6 hours under constant stirring and
temperature. Upon completion, the reaction mixture was cooled to room temperature
and the resulting solid products were isolated by centrifugation, followed by
repeated washing with ethanol and DI to remove residual surfactants and
unreacted species. The purified solids were finally redispersed in DI for
further analysis. The morphology and structural features of the synthesized
particles were characterized using scanning electron microscopy (SEM) and
transmission electron microscopy (TEM).
Results
Solvent effect on particle interaction
Substituting ethanol with n-butanol fundamentally alters the reaction environment governing silica particle formation from TEOS. The lower polarity and dielectric constant of n-butanol reduce the rate of TEOS hydrolysis and subsequent condensation, thereby slowing nucleation kinetics and extending the growth window. In addition, diminished dielectric screening weakens electrostatic stabilization around silica particles, leading to a reduced magnitude of zeta potential3. As a result, particles experience less immediate electrostatic repulsion upon close approach. The higher viscosity of n-butanol further increases the residence time during particle-particle encounters, allowing sufficient interfacial contact for silanol groups (Si-OH) to undergo localized condensation4. Collectively, these solvent-mediated effects shift the system from a regime dominated by rapid charge-stabilized separation toward one favouring controlled interparticle fusion, thereby promoting the formation of stable doublet and triplet structures rather than isolated monodisperse spheres.
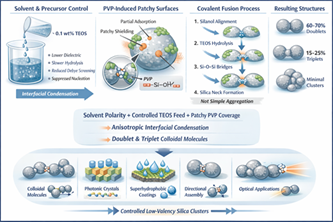
Figure 1: Schematic diagram of PVP controlled anisotropy work
Influence of PVP concentration
The concentration
and molecular weight of PVP played a decisive role in directing the morphology
of the resulting silica assemblies. The observed structural evolution from
isolated spheres to doublets, triplets and eventually irregular aggregates can
be rationalized by considering polymer adsorption behavior, steric
stabilization and interparticle bridging mechanisms5.
At very low PVP concentrations, the silica surface remained largely exposed and polymer adsorption was insufficient to modify interparticle interaction anisotropically. Under these conditions, electrostatic stabilization dominated and particles either remained as isolated monodisperse spheres or underwent limited uncontrolled aggregation depending on collision frequency. The absence of selective surface masking prevented directional bonding6.
At moderate concentration and molecular weight (≈40 kDa), discrete doublets became the dominant morphology. In this regime, PVP adsorbed partially and non-uniformly onto the silica surface through hydrogen bonding between carbonyl groups of the polymer and surface silanol (Si–OH) groups. Importantly, adsorption did not produce complete steric shielding. Instead, it generated patchy surface coverage, leaving localized reactive domains exposed. During particle–particle collisions, these uncovered regions enabled silanol condensation and formation of permanent Si-O-Si bridges7. Because polymer coverage was incomplete yet sufficient to suppress random aggregation, directional fusion predominantly resulted in doublets rather than larger clusters.
When higher
molecular weight PVP or higher concentrations were employed, the probability of
triplet formation increased. Longer polymer chains extended further into
solution and were capable of simultaneously interacting with multiple
particles, introducing a mild bridging flocculation effect. This enhanced
collision efficiency and increased the likelihood of a third particle attaching
to an existing doublet. Additionally, higher surface coverage altered steric
repulsion asymmetrically, further promoting anisotropic cluster growth.
However, careful balance was required to avoid runaway aggregation8.
Under excessive PVP conditions, two competing effects emerged. In some cases, dense polymer adsorption provided strong steric stabilization, preventing fusion and yielding predominantly isolated particles. In other instances, particularly at high molecular weight polymer chain entanglement induced non-specific bridging, resulting in random aggregates rather than well-defined doublets or triplets. Thus, excessive polymer disrupted the delicate kinetic balance required for controlled neck formation.
Overall, the results demonstrate that optimal PVP concentration creates a regime of partial surface coverage where steric stabilization and reactive exposure coexist. This intermediate state generates anisotropic reactive domains that enable directional condensation-driven bonding, thereby facilitating the formation of well-defined colloidal doublets and triplets instead of isotropic or randomly aggregated structures. Clearly the demonstrations in (Figures 2).

Figure 2: Images showcasing doublets and triplets colloidal molecules
Role of TEOS concentration in colloidal molecule
formation
The controlled use
of 0.1 wt% Tetraethyl orthosilicate (TEOS) in both the primary nucleation stage
and the secondary feed played a central role in directing the formation of doublet
and triplet silica structures. The relatively low precursor concentration
ensured that silica growth proceeded under kinetically moderated conditions
rather than rapid homogeneous nucleation.
During the initial
addition of 0.1 wt% TEOS into the n-butanol–ammonium hydroxide medium,
hydrolysis and condensation occurred gradually. The onset of turbidity
indicated controlled nucleation of primary silica seeds. Because the TEOS
concentration was limited, supersaturation levels remained modest, resulting in
a reduced nucleation burst and favouring uniform particle growth rather than
secondary particle formation.
Introduction of
0.05 wt% PVP after turbidity onset ensured that polymer adsorption occurred
predominantly on pre-formed silica surfaces rather than interfering with
nucleation kinetics. At this stage, particles possessed reactive silanol groups
available for further condensation.
The secondary
controlled feed of TEOS at the same low concentration (0.1 wt%) was
particularly critical for colloidal molecule formation. Because the
concentration was not high enough to trigger a new nucleation event, the
freshly hydrolyzed silicate species preferentially condensed onto existing
silica surfaces. When particles collided-facilitated by reduced electrostatic
stabilization in n-butanol-the local concentration of silanol groups at contact
interfaces became elevated. The slow supply of TEOS enabled localized
interfacial condensation, resulting in neck formation rather than independent
growth.
If TEOS concentration
had been higher, two competing phenomena would likely dominate:
·
Secondary
nucleation, producing new smaller particles rather than promoting fusion.
·
Rapid isotropic
shell growth, thickening individual particles and reducing probability of
directional bonding.
Conversely, if
TEOS concentration were significantly lower, insufficient silicate species
would be available to reinforce particle contacts and collisions would remain
reversible, preventing permanent doublet or triplet formation.
Thus, maintaining
TEOS at 0.1 wt% in both stages created a kinetic regime in which:
· Nucleation was controlled and limited.
· Growth occurred preferentially on existing particles.
· Interparticle contacts were chemically stabilized via
condensation.
· Cluster size remained restricted to doublets and
triplets rather than higher aggregates.
The overnight
aging period further allowed gradual condensation at contact points,
strengthening Si-O-Si bridges and stabilizing the colloidal molecule
architecture. Overall, the controlled low TEOS concentration functioned as a
key parameter in shifting the system from homogeneous particle growth toward
anisotropic, condensation-driven colloidal cluster formation.
Morphology distribution
Under optimized
reaction conditions-specifically controlled TEOS feed (0.1 wt%), moderate 40
kDa Polyvinylpyrrolidone concentration (0.05 wt%) and regulated ammonia content-the
system exhibited a highly selective formation of anisotropic silica clusters.
Quantitative morphological analysis from SEM micrographs revealed that
approximately 60-70% of the population consisted of doublets, while 15–25%
formed triplets, with only a negligible fraction of higher-order aggregates.
The predominance
of doublets indicates that the kinetic window favored single fusion events
between two particles9. This
suggests that collision frequency and surface reactivity were sufficiently
balanced to promote one stable interfacial condensation event before steric
stabilization and reduced mobility limited further aggregation. Once a doublet
formed, partial PVP coverage and increasing steric hindrance around the fused
structure likely decreased the probability of additional particle attachment,
thereby limiting uncontrolled cluster growth. Triplet formation occurred at a
lower but significant frequency. Importantly, most triplets exhibited linear
configurations, rather than triangular or symmetric arrangements. SEM images of
doublets and triplets are shown in (Figure 2). This structural
preference strongly supports a sequential attachment mechanism rather than
simultaneous three-body fusion. In this process, an initially formed doublet
retains one reactive, partially unshielded domain. A third particle
subsequently collides and condenses at this exposed interface, forming a
chain-like structure (sphere-sphere-sphere). The likelihood of three particles
simultaneously colliding and condensing symmetrically is statistically low in
dilute colloidal systems, further reinforcing the sequential growth model. The
minimal presence of higher-order aggregates indicates that steric stabilization
from adsorbed PVP and the low TEOS concentration effectively suppressed runaway
cluster formation. As clusters grow, their hydrodynamic size increases,
diffusion slows and steric barriers become more pronounced, reducing additional
attachment probability. Consequently, the system self-limits at low valency
structures, primarily yielding doublets and some triplets.
In a moderately
polar solvent such as n-butanol, PVP adsorbed non-uniformly on TPM-functionalized
silica spheres can act as a nanoscale capillary bridge that locally
concentrates hydrolyzed species derived from TEOS. Due to its carbonyl
functionality and flexible chain architecture, PVP forms hydrogen-bonding
interactions with surface silanols while simultaneously retaining polar
silicate oligomers within polymer-rich microdomains, especially at
particle–particle contact points or surface irregularities. In the lower
dielectric environment of n-butanol, polymer chains partially collapse and
increase local viscosity, reducing precursor diffusion and creating confined
regions of enhanced supersaturation. This localized enrichment promotes
preferential siloxane condensation (Si-O-Si) at specific interfacial sites
rather than uniform radial shell growth, thereby directing anisotropic silica
deposition and reinforcing neck formation between TPM spheres. Thus, beyond
steric stabilization, PVP functions as a transient capillary microreactor that
spatially biases TEOS condensation and enables controlled directional growth.
Mechanistic
Discussion
Condensation-driven neck growth
When silica
particles bearing partially adsorbed PVP collide in the butanol-based reaction
medium, their interaction is governed not only by physical contact but by localized
surface chemistry. Because PVP coverage is incomplete at optimized
concentrations, certain regions of the silica surface remain exposed,
particularly silanol (Si-OH) groups that are highly reactive under basic
conditions.
Upon collision,
these exposed silanol groups from two approaching particles come into close
proximity and align at the contact interface. The reduced electrostatic
repulsion in n-butanol allows sufficient residence time for molecular
reorganization at this junction clearly indiated in (Figure 3).
Simultaneously, the secondary feed of hydrolyzed TEOS supplies reactive
silicate species into the surrounding medium. Because particle surfaces provide
energetically favourable condensation sites, hydrolysis and condensation
reactions become locally concentrated at the interface between touching
particles.
This localized
reaction leads to the formation of siloxane (Si-O-Si) bridges, progressively
converting reversible physical contact into irreversible chemical fusion. As
condensation continues, a solid silica neck develops, structurally integrating
the two particles into a stable doublet. If an additional collision occurs at
an exposed reactive domain of a pre-formed doublet, the same mechanism enables
triplet formation through sequential attachment.
This process is fundamentally distinct from simple aggregation. In physical aggregation, particles are held together by weak van der Waals or electrostatic interactions and can be redispersed under mild perturbation. In contrast, the present mechanism involves covalent siloxane bond formation, permanently fusing particles into a unified structure. The resulting neck region exhibits continuous silica framework growth, confirming that cluster formation arises from condensation-driven interfacial polymerization rather than reversible colloidal flocculation which is clearly demonstrated in (Figure 3).
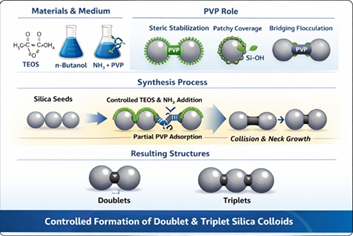
Figure 3: Intermediate transitions during PVP based controlled anisotropy
Role of solvent polarity
The solvent
environment plays a fundamental physicochemical role in dictating colloidal
interaction forces and growth pathways. When the reaction medium is shifted
from ethanol to n-butanol, the dielectric constant decreases significantly. In
colloidal systems, the dielectric constant directly influences the thickness
and effectiveness of the electrical double layer surrounding charged particles.
Under basic conditions, silica particles possess negatively charged siloxide
(Si-O⁻) groups on their surfaces, which generate electrostatic repulsion
between approaching particles.
In a higher
dielectric solvent such as ethanol, charge stabilization is strong and the
Debye screening length is effectively extended, meaning that electrostatic
repulsion acts over a longer range. As a result, particles rarely approach
closely enough for surface silanol groups to interact chemically. Collisions
are brief and largely reversible, favoring monodisperse spherical growth.
In contrast,
n-butanol has a lower dielectric constant, which compresses the electrical
double layer and reduces Debye screening efficiency10. Consequently, the magnitude and effective range of
electrostatic repulsion decrease. Particles can approach more closely before
repulsive forces dominate, increasing the probability of intimate surface
contact. This close approach is critical because condensation reactions between
silanol groups require molecular-level proximity.
Simultaneously,
hydrolysis of Tetraethyl orthosilicate (TEOS) proceeds more slowly in n-butanol
due to reduced solvent polarity and altered solvation of reactants. Slower
hydrolysis leads to lower supersaturation of silicate species in solution. This
suppresses homogeneous nucleation of new particles, which would otherwise compete
with cluster formation. Instead of forming independent nuclei, hydrolysed
silicate species preferentially condense on energetically favourable sites-namely,
existing silica surfaces and particularly particle–particle contact interfaces.
Thus, two synergistic
effects occur:
·
Reduced
electrostatic repulsion enables particles to remain in contact long enough for
chemical reactions to occur.
·
Slower hydrolysis
minimizes new particle formation and channels silica growth toward interfacial
condensation.
Together, these
solvent-induced changes redirect the reaction pathway from isotropic particle
nucleation toward anisotropic, interface-driven fusion. The result is a system
that favours the formation of chemically bonded doublets and triplets rather
than isolated monodisperse spheres.
Polymer-mediated anisotropic shielding
The adsorption
behavior of Polyvinylpyrrolidone (PVP) on silica surfaces is inherently dynamic
and spatially non-uniform. PVP interacts with silica primarily through hydrogen
bonding between its lactam carbonyl groups and surface silanol (Si-OH)
functionalities. However, this interaction is not equivalent across the entire
particle surface. Factors such as local curvature, silanol density, polymer
chain conformation and solvent quality influence how and where the polymer
adsorbs.
At moderate
concentrations, PVP chains adopt loop-and-tail configurations rather than
forming a dense, continuous layer. Some segments anchor to the surface, while
other portions extend into the surrounding solvent. Because adsorption occurs
through multiple weak, reversible interactions, surface coverage remains
incomplete and heterogeneous. This results in “patchy” particles-regions of the
surface are sterically shielded by polymer, while other domains remain exposed
and chemically reactive.
These exposed
silanol-rich domains act as localized reactive sites during particle–particle
encounters. When two partially shielded particles collide, condensation is more
likely to occur between unprotected regions, while polymer-covered regions
resist further interaction. This spatial asymmetry effectively mimics
directional bonding seen in molecular systems, where specific reactive sites
dictate bonding geometry. As a result, fusion events become anisotropic rather
than isotropic, favouring controlled doublet and triplet formation instead of
random aggregation.
When higher
molecular weight PVP is used, the polymer chains are longer and possess larger
hydrodynamic radii. Such chains can extend far enough into solution to
simultaneously interact with two neighbouring particles. This introduces a
secondary mechanism known as mild bridging flocculation. In this scenario, a
single polymer chain transiently binds multiple particles, increasing their
probability of remaining in close proximity. While still moderated by steric
stabilization, this effect enhances the likelihood that a third particle
attaches to a pre-formed doublet, thereby increasing triplet formation.
Importantly, the
bridging induced by high molecular weight PVP is subtle under optimized
conditions. It promotes sequential attachment without causing uncontrolled
clustering. Thus, polymer-mediated anisotropic shielding and controlled
bridging together regulate particle valency, enabling selective formation of
low-order colloidal molecules.
Anistopic
Organosilica Colloidal Molecule
The formation of raspberry-like and buckle-hole colloidal molecules in this system shown in (Figure 4) can be understood as a consequence of the dynamic evolution of a double-emulsion template coupled with interfacial organosilica condensation and polymerization. Initially, the precursor mixture containing 3-(Trimethoxysilyl) propyl methacrylate and water, stabilized by Span 80 and further dispersed in a butanol phase with Cetyltrimethylammonium bromide, generates a water-in-oil-in-water (W/O/W) double emulsion, as evidenced by the optical micrograph showing multiple internal droplets confined within a larger parent droplet. These interfaces act as highly active reaction zones where, under basic conditions provided by ammonium hydroxide, TPM undergoes hydrolysis to form silanol groups followed by condensation into an organosilica network11. Due to the curvature and compositional heterogeneity of the emulsion, condensation does not occur uniformly; instead, it is localized at regions of high interfacial curvature and at contact points between internal droplets and the outer interface. As the reaction proceeds at elevated temperature, internal aqueous droplets undergo coalescence, shrinkage and migration driven by interfacial tension gradients and solvent exchange between butanol and water, generating Marangoni flows and transient concentration gradients of TPM oligomers12. These dynamic processes create anisotropic growth conditions, leading to the heterogeneous nucleation of secondary organosilica domains on the surface of primary particles, which manifests as the characteristic raspberry-like morphology observed in SEM. Simultaneously, partial encapsulation followed by collapse or escape of internal droplets results in localized voids or indentations; because the surrounding organosilica network has already begun to solidify, these deformations cannot relax, giving rise to buckle-hole structures. The subsequent addition of KPS initiates free-radical polymerization of the methacrylate groups within TPM13, forming a crosslinked hybrid network that kinetically arrests the evolving morphology and preserves these non-equilibrium features. Thus, the final structures arise from a complex interplay of double-emulsion templating, interfacial condensation, surfactant-mediated curvature stabilization and polymerization-driven fixation, resulting in anisotropic colloidal molecules with hierarchical surface and internal architectures.
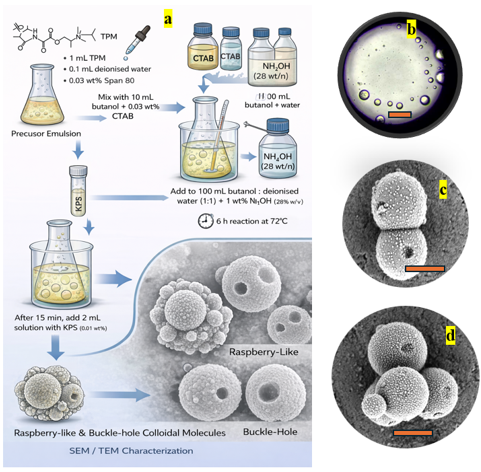
Figure 4: (a) Overall procedure of Anisotropic functionality of organosilica.
(b) Microscopic image of W/O/W emulsion of precursor solution. (c) Raspberry
buckled colloidal doublet. (d) Raspberry buckled colloidal quadruplet. Scale
bar is 500nm
Applications
The synthesized
doublet and triplet silica colloids possess intrinsic anisotropy that
significantly expands their functional utility compared to conventional
spherical particles. Because these structures consist of chemically fused lobes
connected by a rigid silica neck, they behave as low-valency “colloidal
molecules,” where each lobe acts as a structural unit analogous to atoms in
molecular systems. This directional geometry enables predictable bonding
orientations during assembly, making them highly suitable for colloidal
molecule assembly. Unlike isotropic spheres that pack randomly, doublets and
triplets can organize into ordered architectures with defined angles and
connectivity, enabling programmable mesoscale structures.
As photonic
crystal precursors, these anisotropic colloids offer additional advantages.
Photonic materials rely on periodic variations in refractive index to
manipulate light propagation. The fused multi-lobed geometry introduces
symmetry breaking and directional periodicity into assembled lattices. This can
result in modified photonic band structures, anisotropic scattering behavior
and polarization-dependent optical responses. Because silica has good optical
transparency and chemical stability, such colloidal molecules can serve as
building blocks for advanced photonic metamaterials.
For directional
self-assembly platforms, these structures provide controlled valency. A doublet
has two principal interaction domains, while a triplet can present three
directional interaction points. Such geometry-dependent bonding promotes the
formation of chains, branched networks or specific lattice arrangements under
external fields or surface confinement. This makes them attractive for studying
controlled assembly kinetics and emergent structural order.
Finally, in
optical scattering studies, anisotropic silica clusters exhibit different
light-scattering behavior compared to spheres of equivalent volume. The
presence of multiple lobes alters scattering cross-sections, angular
distribution and interference effects. This enables investigation of
anisotropic Mie scattering, multiple scattering pathways and enhanced
light–matter interaction phenomena.
Overall, the
anisotropic geometry of doublet and triplet silica colloids enables controlled
packing behavior, tunable interaction valency and enhanced optical
functionality. These features position them as versatile building blocks for
advanced materials engineering and fundamental colloidal science research.
Conclusion
A modified Wilhelm
Stöber-type synthesis conducted in n-butanol with controlled
Polyvinylpyrrolidone adsorption enables selective formation of doublet and
triplet silica colloids from Tetraethyl orthosilicate. Solvent polarity
modulation slows hydrolysis and reduces electrostatic stabilization, while
partial polymer coverage generates anisotropic bonding sites. Controlled
secondary TEOS feed promotes condensation-driven neck formation, yielding
discrete colloidal clusters.
This scalable solution-based approach provides a versatile route toward colloidal molecules with tunable valency and structural precision, expanding the synthetic toolbox for advanced materials design.
References
4. Anabel
RE, Caregnato P, Arce VB, et al. Synthesis and Characterization of Butoxylated
Silica Nanoparticles. Reaction with Benzophenone Triplet States. J Physical Chem 2007;111(21):7623-7628.
13. Akbar A, Kaykhaii M. Kinetics and Mechanism of Potassium Persulphate/L-Serine Initiated Polymerization of Methylmethacrylate. J Polymer Res 2004;11(3):231-238.